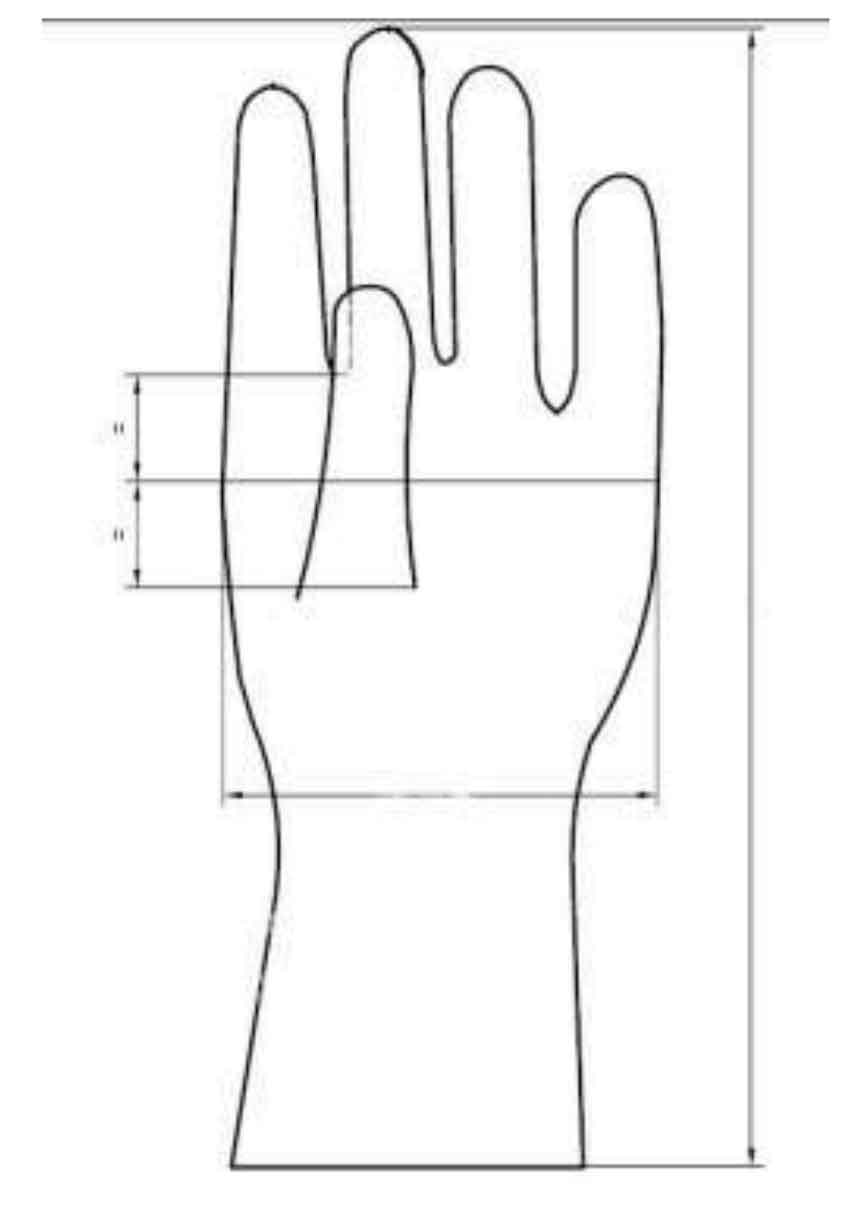
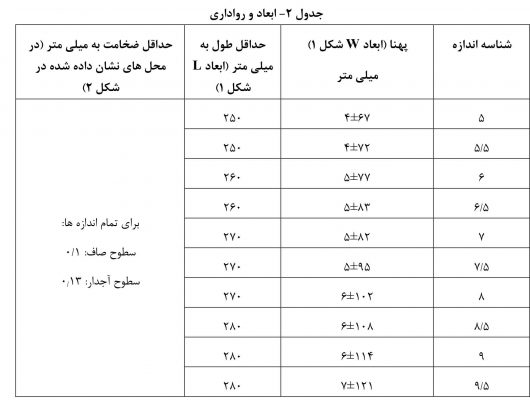
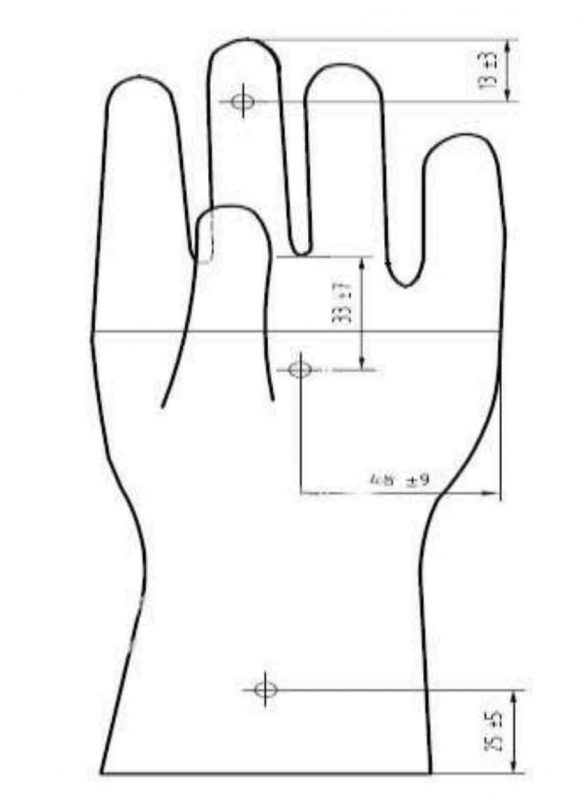
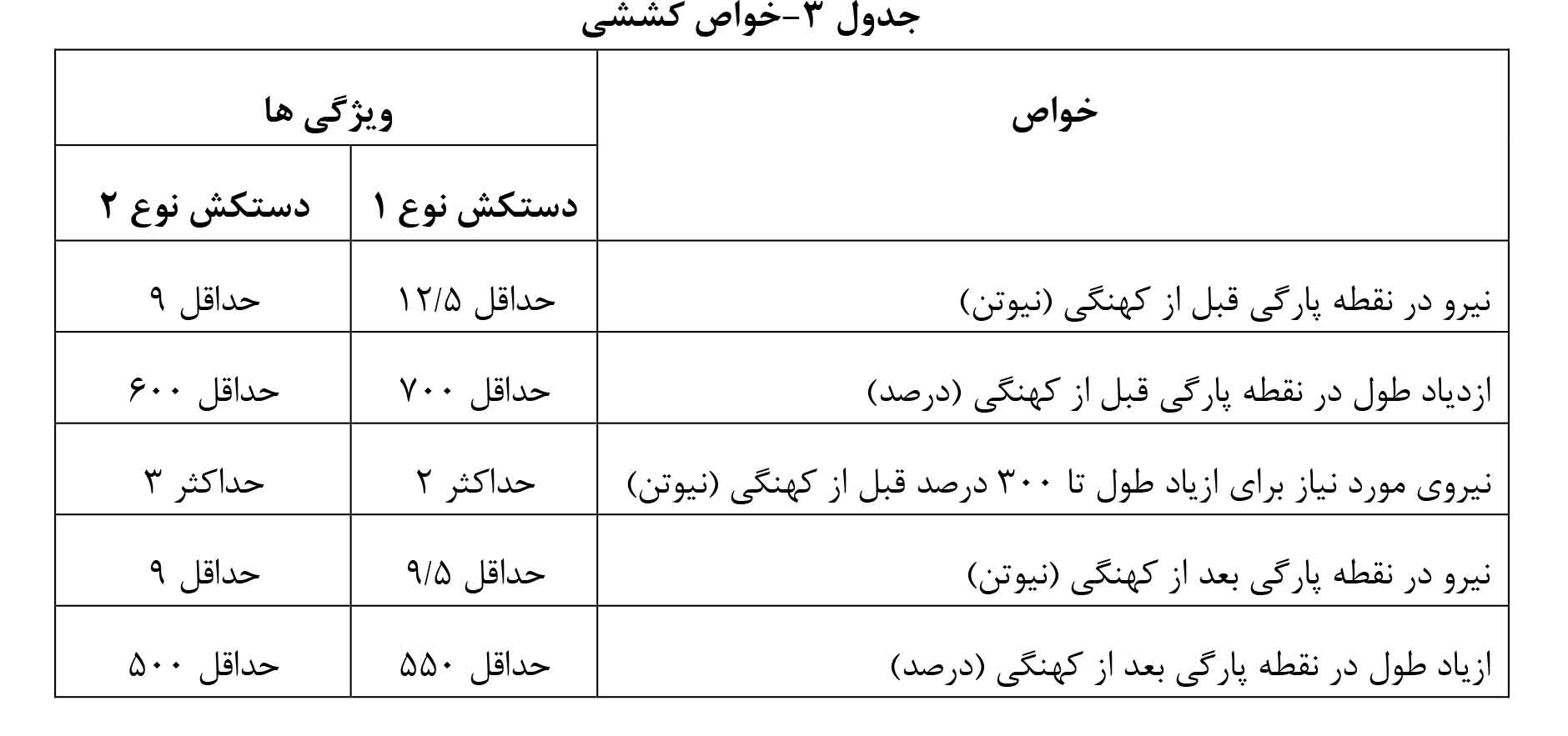
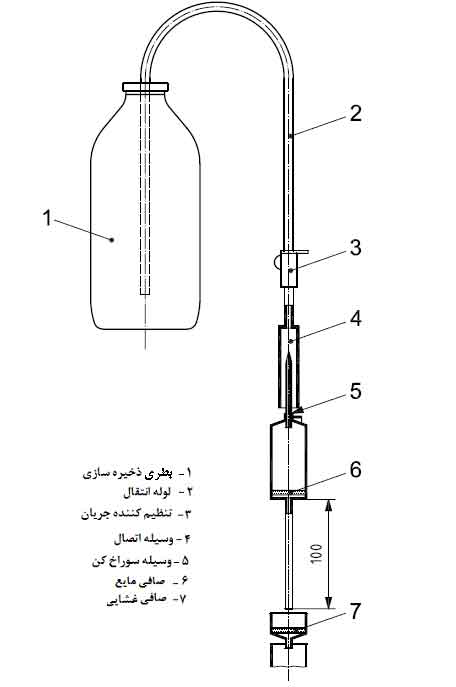
استاندارد ایزو ۱۳۴۸۵
ویرایش ۲۰۱۶
ترجمه به فارسی توسط شرکت QS سوئیس
مترجم: دکتر علی محمد پور بازنگری: فرداد اسلامی
۱-۰ کلیات
این استاندارد بین المللی الزاماتی را برای سیستم مدیریت کیفیت مشخص می کند که می تواند از سوی یک سازمان فعال در یک یا چند مرحله از چرخه ی عمر یک تجهیز پزشکی شامل طراحی و تکوین, تولید, انبارش و توزیع ، نصب یا ارائه خدمات مربوطه (مانند پشتیبانی فنی) ، انهدام نهائی تجهیز پزشکی و طراحی و تکوین یا ارائه خدمات مرتبط مورد استفاده قرار گیرد. هم چنین، این استاندارد می تواند از سوی تامین کنندگان یا دیگر طرف های بیرونی ارائه دهنده محصول (مانند مواد اولیه، اجزا، زیرمجموعه ها، تجهیزات پزشکی، خدمات سترونی، خدمات کالیبراسیون، خدمات توزیع، خدمات نگهداری به این گونه سازمان ها، مورد استفاده قرار گیرد. تامین کننده یا طرف بیرونی می تواند داوطلبانه انتخاب نماید که با الزامات این استاندارد بین المللی انطباق داشته باشد یا از طریق قرارداد، ملزم به انطباق شود.
چندین حوزه قانونی ، دارای الزامات مقرراتی هستند که در سیستم مدیریت کیفیت سازمان ها با نقش های متنوع در زنجیره تامین تجهیزات پزشکی، کاربرد دارند. در نتیجه، این استاندراد بین المللی انتظار دارد که سازمان:
- نقش خودش را تحت الزامات قانونی اجرائی، شناسایی کند،
- الزامات مقرراتی را که در فعالیت های تحت این نقش ها کاربرد دارد، شناسایی کند،
- این الزامات قانونی اجرائی را درون سیستم مدیریت خود لحاظ نماید.
تعاریف در الزامات قانونی اجرائی ، کشور به کشور و منطقه منطقه، متفاوت است. سازمان نیازمند آن است تا درک کند چگونه تعاریف این استاندارد بین المللی تحت تعاریف مقرراتی، در حوزه های قانونی که تجهیزات پزشکی عرضه میگردد، تفسیر خواهد شد.
همچنین، این استاندارد می تواند از سوی طرف های درونی و بیرونی شامل موسسات گواهی دهنده برای ارزیابی توانایی سازمان در برآورده سازی الزامات مشتری و الزامات قانونی اجرائی در سیستم مدیریت کیفیت و الزامات سازمان مورد استفاده قرار گیرد. تاکید می شود که الزامات سیستم مدیریت کیفیت مشخص شده در این استاندارد بین المللی، تکمیل کننده الزامات فنی محصول می باشند و برای برآورده سازی الزامات مشتری و الزامات قانونی اجرائی جهت ایمنی و عملکرد، ضروری هستند.
پذیرش سیستم مدیریت کیفیت, یک تصمیم راهبردی برای سازمان است. طراحی و اجرای سیستم مدیریت کیفیت در یک سازمان, تحت تاثیر موارد زیر قرار می گیرد
أ) محیط سازمانی، تغییرات در آن محیط، و تاثیری که محیط سازمانی بر انطباق تجهیزات پزشکی دارد
ب) نیازهای متغیر سازمان پ) اهداف ویژه سازمان ت) محصولی که سازمان ارائه می کند ث) فرایندهایی که سازمان در اختیار دارد ج) اندازه سازمان و ساختار سازمانی چ الزامات مقرراتی قابل کاربرد در فعالیتهای سازمان
هدف این استاندارد بین المللی، یکسان سازی ساختار سیستم های مدیریت کیفیت مختلف، مستند سازی یکسان و یا مستندسازی هم راستا با ساختار بندی این استاندارد بین المللی ، نمی باشد.
تعداد و تنوع تجهیزات پزشکی بسیار وسیع بوده و برخی الزامات خاص این استاندارد بین المللی فقط به گروه های نامبرده شده تجهیزات پزشکی مربوط می باشند. این گروه ها در بند ۳ تعریف شده اند.
۲-۰ شفاف سازی مفاهیم
در متن این استاندارد بین المللی، واژگان یا عباراتی که در زیر توصیف شده اند، مورد استفاده قرار می گیرند.
و هنگامی که الزامی از طریق عبارت در موارد مقتضی» مطرح می گردد، مقتضی تلقی می شود مگر این که سازمان بتواند خلاف آنرا توجیه نماید. یک الزام در صورتیکه برای موارد زیر ضروری باشد، مقتضی در نظر گرفته میشود :
- برآورده سازی الزامات توسط محصول • انطباق با الزامات مقرراتی قابل کاربرد
- انجام اقدام اصلاحی توسط سازمان • مدیریت کردن ریسک ها توسط سازمان
هنگامی که واژه «ریسک» به کار می رود، کاربرد این واژه در دامنه شمول این استاندارد بین المللی، به الزامات ایمنی یا عملکردی تجهیزات پزشکی یا برآورده سازی الزامات قانونی اجرائی، مربوط میشود.
- هنگامی که لازم است یک الزام مدون شود ، علاوه بر مستند سازی میبایست استقرار اجرا و نگهداری نیز گردد .
و هنگامی که واژه «محصول» استفاده می شود، می تواند معنی «خدمت» را نیز بدهد. محصول بعنوان برونداد یا الزام مورد نظر برای یک مشتری یاهر برونداد حاصل از فرایند پدیدآوری محصول، بکار میرود .
- هنگامی که واژه «الزامات مقرراتی» استفاده می شود، در برگیرنده الزامات هر قانون قابل کاربرد برای کاربر این استاندارد بین المللی می شود (از قبیل مقررات ، آئین نامه ها، احکام یا دستورالعمل ها). کاربرد واژه «الزامات مقرراتی» به الزامات سیستم مدیریت کیفیت و ایمنی یا عملکرد تجهیز پزشکی محدود میگردد .
در این استاندارد بین المللی، عناوین لفظی زیر استفاده می شود:
- باید : نشاندهنده یک الزام است.| • بایستی : نشاندهنده یک توصیه است.| • امکان پذیر است: نشاندهنده یک اجازه
است.
می تواند، نشاندهنده یک احتمال یا قابلیت است.
اطلاعاتی که بعنوان «یادآوری » مشخص شده است، یک راهنما جهت درک یا شفاف سازی الزام مربوطه می باشد.
۳-۰ رویکرد فرایندی
این استاندارد بین المللی مبتنی بر رویکرد فرایندی در مدیریت کیفیت می باشد. هر فعالیتی که دروندادی را به بروندادی تبدیل نماید، می تواند به عنوان یک فرایند در نظر گرفته شود. غالبا، بروندادهای یک فرایند مستقیما در وندادهای فرایند بعدی را تشکیل می دهند.
برای اینکه سازمان موثر عمل نماید, نیاز دارد فرآیندهای بهم مرتبط متعددی را شناسایی و مدیریت نماید. بکارگیری سیستمی از فرآیندها در درون یک سازمان ، همراه با مشخص کردن و تعامل این فرآیندها و مدیریت کردن آنها به منظور دستیابی به نتایج مطلوب “ رویکرد فرآیندی“ نامیده می شود.
این رویکرد هنگامی که در سیستم مدیریت کیفیت بکار گرفته می شود، بر اهمیت موارد زیر تاکید مینماید: و درک و برآورده سازی الزامات. و در نظر گرفتن فرایندها به لحاظ ارزش افزائی و حصول نتایج عملکرد و اثربخشی فرایندها. و بهبود فرایندها بر پایه اندازه گیری عینی.
۴-۰ارتباط با استاندارد 9001 ISO در حالی که این استاندارد مستقل می باشد اساس آن بر پایه استاندارد
9001:2008 بوده که با استاندارد ISO9001:2015 جایگزین شده است. برای راحتی کاربران پیوست ب) مشابهت میان این استاندارد بین المللی و ISO9001:2015 را نشان می دهد.
این استاندارد بین المللی قصد دارد همراستایی جهانی در خصوص الزامات مقرراتی مقتضی برای سیستم مدیریت کیفیت بکار گرفته شده در سازمان های فعال در یک یا چند مرحله از چرخه عمر یک تجهیز پزشکی را، تسهیل کند. این استاندارد بین المللی شامل الزامات ویژه برای سازمان های فعال در چرخه عمر تجهیزات پزشکی می باشد و برخی از الزامات ISO9001 را که به عنوان الزامات مقرراتی، مقتضی نیستند، حذف نموده است. به همین دلیل، سازمان هایی که سیستم مدیریت کیفیت آنها با این استاندارد بین المللی انطباق دارد ، نمی توانند مدعی انطباق با ISO9001 باشند مگر این که سیستم مدیریت کیفیت آنها، تمامی الزامات ISO9001 را برآورده نماید.
۵۰سازگاری با دیگر سیستمهای مدیریتی این استاندارد در برگیرنده الزامات خاص سایر سیستم های مدیریت از قبیل الزاماتی که در مدیریت زیست محیطی، مدیریت بهداشت و ایمنی کار یا مدیریت مالی در نظر گرفته شده اند، نمی باشد.
به هر حال این استاندارد بین المللی سازمان را قادر می سازد که سیستم مدیریت خود را با الزامات سیستم های مدیریت ذیربط , همراستا یا یکپارچه نماید. برای یک سازمان این امکان وجود دارد که سیستم مدیریت فعلی خود را به منظور استقرار یک سیستم مدیریت کیفیت بر طبق الزامات این استاندارد بین المللی , تطبيق دهد.
1- دامنه این استاندارد بین المللی، الزامات یک سیستم مدیریت کیفیت را در مواردی مشخص می کند که سازمان به اثبات توانایی خود در ارایه تجهیزات پزشکی و خدمات مرتبط که بطور مستمر خواسته های مشتری و الزامات قانونی اجرائی مربوط به آن را برآورده می نماید، نیاز دارد.
این گونه سازمان ها می توانند در یک یا چند مرحله از چرخه عمر، شامل طراحی و تکوین، تولید، انبارش و توزیع، نصب یا خدمات دهی به یک تجهیز پزشکی یا طراحی و تکوین یا ارائه فعالیت های مرتبط (مانند پشتیبانی فنی) فعال باشند. همچنین تامین کنندگان و طرف های بیرونی که محصولاتی شامل خدمات مرتبط با سیستم مدیریت کیفیت به این گونه سازمان ها ارائه می دهند نیز می توانند این استاندارد بین المللی را مورد استفاده قرار دهند.
الزامات این استاندارد بین المللی برای سازمان ها صرف نظر از اندازه و نوع شان ، به استثناء مواردی که صراحتا بیان شده است، کاربرد دارد. هر کجا الزاماتی تحت عنوان «بکار گرفته شده در تجهیزات پزشکی» مشخص شده باشد، الزامات برای خدمات مرتبط ارائه شده توسط سازمان نیز کاربرد دارد.
فرآیندهایی که در این استاندارد بین المللی الزامی بوده و برای تجهیزات پزشکی مورد نظر کاربرد دارد ولی توسط سازمان بکار گرفته نمی شوند، جزء مسوولیت سازمان بوده و از طریق پایش، نگهداشت و کنترل این فرایندها در سیستم مدیریت کیفیت سازمان در نظر گرفته می شوند.
چنانچه الزامات مقرراتی اجازه حذف کنترل های طراحی و تکوین را بدهد، این مورد می تواند به عنوان توجیه حذف آنها از سیستم مدیریت کیفیت، بکار گرفته شود. این الزامات مقرراتی می توانند رویکرد جایگزینی را ارائه دهند که در سیستم مدیریت کیفیت اشاره شده است. این مسئولیت سازمان است تا اطمینان یابد ادعاهای انطباق با این استاندارد بین المللی باز گو کننده هرگونه حذف کنترل های طراحی و تکوین می باشد.
در صورتیکه هر الزامی از بندهای ۷۰۶ و یا۸ این استاندارد بین المللی ، بسته به فعالیت های به عهده گرفته شده از جانب سازمان یا ماهیت تجهیز پزشکی که سیستم مدیریت کیفیت برای آن اعمال شده است، کاربرد نداشته باشد، سازمان نیازی به در بر گرفتن آنها در سیستم مدیریت کیفیت خود ندارد. سازمان توجیه هر موردی که غیر قابل کاربرد تعیین شده است را همانگونه که در بندر ۲-۲-۴) اشاره شده ، ثبت می نماید.
۲- مراجع اصلی سند مرجعی که نام آن در زیر می آید ، بطور کلی یا جزئی ، بعنوان مرجع این استاندارد بوده و برای بکارگیری این استاندارد ضروری میباشد. در مورد مراجع تاريخ دار, فقط نسخه ی ذکر شده کاربرد دارد. برای مراجع بدون تاریخ آخرین نسخه ی مرجع (شامل هر گونه اصلاحات کاربرد دارد. استاندارد ISO9000:2005
– سیستم های مدیریت کیفیت – مبانی و واژگان
٣- واژگان و تعاریف در این استاندارد ، واژگان و تعاریف ذکر شده دراستاندارد ISO9000:2005 ، بعلاوه موارد زیر بکار رفته است :
۳-۱ هشدارهای توصیه ای
هشداری که سازمان پس از تحویل تجهیز پزشکی, جهت ارائه اطلاعات تکمیلی یا اضافی و مشاوره در اقدامی که باید انجام گیرد جهت موارد زیر صادر می کند:
- استفاده از تجهیزات پزشکی • تغییر و تبدیل تجهیز پزشکی
- بازگرداندن تجهیز پزشکی به سازمانی که آن را تامین نموده است
- انهدام تجهیز پزشکی
یادآوری: صدور هشدار توصیه ای می تواند جهت انطباق با الزامات قانونی اجرائی، الزامی شده باشد.
۳-۲ نماینده مجاز
شخص حقیقی یا حقوقی در یک کشور یا حوزه قانونی ، که مجوز مکتوبی را از سازنده دریافت کرده است تا از طرف او به عنوان تعهد ثانویه تحت مقررات کشور یا مصوبات حوزه قانونی، فعالیت کند. منبع : GHTF/SGI / NO55.2009.5.2
۳-۳ ارزیابی بالینی
ارزیابی و تحلیل اطلاعات بالینی مربوط به یک تجهیز پزشکی بمنظور تصدیق ایمنی و عملکرد بالینی تجهیز هنگام کاربرد موردنظر سازنده
۳-۴ شکایت
تبادل اطلاعات مکتوب، الکترونیکی یا شفاهی که نواقص مربوط به هویت، کیفیت، دوام، قابلیت اعتماد، قابلیت استفاده، ایمنی یا عملکرد یک تجهیز پزشکی که از سازمان ترخیص شده است یا خدماتی که بر عملکرد این گونه تجهیزات پزشکی تاثیر گذار است، را ادعا میکند.
یادآوری: این تعریف از «شکایت» با تعریف ارائه شده دراستاندارد ISO9000:2015 تفاوت دارد.
۳-۵ توزیع کننده
شخصی حقیقی یا حقوقی در زنجیره تامین، که با مسئولیت خود، عرضه تجهیز پزشکی به کاربر نهایی را تسهیل مینماید.
یادآوری: بیش از یک توزیع کننده ممکن است در یک زنجیره تامین فعالیت داشته باشند.
یادآوری: افرادی در زنجیره تامین که از طرف سازنده، وارد کننده یا توزیع کننده در فعالیت هایی مانند انبارش و حمل و نقل دخیل هستند، تحت این تعریف، توزیع کننده محسوب نمی شوند.
منبع : GHTF/SGl/N055:2009,5.3
۳-۶ تجهیز پزشکی کاشتنی
تجهیز پزشکی که تنها با مداخله پزشکی یا جراحی قابل برداشتن است و مدنظر است تا از طریق عمل جراحی به طور کلی یا جزیی در داخل بدن انسان یا یکی از منافذ طبیعی وارد شود. و جایگزین یک سطح مخاطی یا سطح چشم شود. | و بعد از عمل دست کم 30 روز باقی بماند. یادآوری: تعریف تجهیز پزشکی کاشتنی دربرگیرنده تجهیز پزشکی کاشتنی فعال نیز می شود.
۳-۷ واردکننده
شخصی حقیقی یا حقوقی در زنجیره تامین نخستین کسی است که تجهیز پزشکی ساخته شده در یک کشور با حوزه قانونی دیگر را ، در یک کشور با حوزه قانونی که می بایست بازاریابی شود، در دسترس قرار می دهد.
۳-۸ برچسب گذاری
برچسب، دستورالعمل استفاده و هر گونه اطلاعات مرتبط با شناسایی، توضیحات فنی، مقاصد موردنظر یا استفاده صحیح از تجهیزات پزشکی ، به غیر از اسناد حمل می باشد. منبع : GHTF/SGI / N70:2011 , Clause4
۳-۹ چرخه ی عمر
تمامی مراحل عمر یک تجهیز پزشکی از مفهوم سازی اولیه تا انهدام و تعیین تکلیف نهایی.
منبع : ISO14971:2007,2,7
۳-۱۰ سازنده
شخص حقیقی یا حقوقی که مسئولیت طراحی و یا ساخت یک تجهیز پزشکی به منظور عرضه آن برای استفاده تحت نام خود را بعهده دارد، چه این تجهیز پزشکی را خود او ، یاشخص دیگری با مسئولیت از طرف او ، طراحی کرده و یا ساخته باشد.
یادآوری: این شخص حقیقی یا حقوقی» مسئولیت نهایی اطمینان از تطابق با تمامی الزامات قانونی اجرائی برای تجهیز پزشکی در کشورها یا حوزه های قانونی ای که قرار است در آن عرضه شده یا فروخته شود را به عهده دارد مگر این که این مسئولیت از طرف نهاد ذیصلاح مقرراتی در درون آن حوزه قانونی ، به شخصی دیگری واگذار شده باشد.
یادآوری: مسئولیت سازنده در اسناد راهنمای GHTF توضیح داده شده است. این مسئولیت ها شامل برآورده سازی الزامات پیش – بازار و پس-بازار مانند گزارش دهی رویدادهای نامطلوب و اطلاع رسانی اقدامات اصلاحی می باشد.
* یادآوری: «طراحی و یاساخت» که در تعریف بالا به آن ارجاع شد، ممکن است دربرگیرنده ایجاد ویژگی های تولید، ساخت، مونتاژ، فرآوری، بسته بندی، بسته بندی مجدد، برچسب گذاری، برچسب گذاری مجدد، سترون سازی، نصب، یاساخت مجدد یک تجهیز پزشکی یا در کنار هم قرار دادن تجهیزات و یا احتمالا دیگر محصولات برای یک مقصود پزشکی باشد.
یادآوری: هر فردی که یک تجهیز پزشکی را که پیشتر، فرد دیگری برای بیمار به خصوصی در مطابقت با دستورالعمل استفاده، تامین کرده بود، مونتاژ یا جفت و جور نماید، سازنده محسوب نمی شود . ارائه مونتاژ یا جفت و جورسازی ، حیطه کاربرد تجهیز پزشکی را تغییر نمی دهد.
یادآوری : هر فردی که حیطه کاربرد یک تجهیز پزشکی را تغییر دهد یا اصلاح نماید، بدون اینکه این تغییر از جانب سازنده اصلی باشد و آن تجهیز را تحت نام خود برای استفاده عرضه نماید، می بایست به عنوان سازنده تجهیز پزشکی تغییر یافته، در نظر گرفته شود.
یادآوری: نماینده مجاز، توزیع کننده یا وارد کننده که تنها نشانی و جزییات تماس خود را به تجهیز پزشکی یا بسته بندی آن اضافه مینماید، بدون اینکه برچسب های موجود تجهیز پزشکی را بپوشاند یا تغییر دهد، سازنده تلقی نمی شود.
یادآوری: در محدوده ای که یک قطعه جانبی در الزامات مقرراتی یک تجهیز پزشکی موضوعیت داشته باشد، فرد مسئول برای طراحی و یاساخت آن قطعه سازنده در نظر گرفته می شود. منبع : GHTF/SG1 / N055:2009,5.1
۳-۱۱ تجهیز پزشکی
وسایل، ادوات، ابزار، ماشین، لوازم، قطعات کار گذاشتنی ، معرف های تشخیصی ، نرم افزار، مواد یا دیگر موارد مشابه یا مرتبط که توسط سازنده در نظر گرفته شده، به تنهایی یا بصورت ترکیبی برای سلامتی انسان برای یک یا چند مقصود پزشکی مشخص زیر، استفاده شود:
- تشخیص, پیشگیری, پایش, معالجه یا تسکین بیماری
- تشخیص, پایش, معالجه, تسکین یا ترمیم آسیب دیدگی
- تحقيق, جایگزینی, اصلاح یا پشتیبانی ساختمان بدن ( Anatomy )یایک فرایند فیزیولوژیک و حمایت و حفظ حیات کنترل بارداری
- ضدعفونی تجهیزات پزشکی
- آماده کردن اطلاعات به وسیله آزمایش تشخیصی نمونه های به دست آمده از بدن انسان و عملیات اصلی مورد نظر خود را در داخل یا خارج از بدن انسان از طریق وسایل و ابزارهای داروشناسی, ایمنی شناسی یا متابولیک به انجام نمی رساند بلکه ممکن است در کارکرد موردنظرش از این طریق، یاری داده شود. |
یادآوری: محصولاتی که ممکن است در برخی حوزه هایی قانونی، تجهیز پزشکی تلقی شوند ، شامل موارد زیر است:
مواد ضد عفونی و وسایل کمکی برای معلولان و تجهیزات جا داده شده در بافت جانوران و یا انسان و تجهیزات برای لقاح آزمایشگاهی و فناوری های کمک به تولید مثل. منبع : GHTF/SGI / NO71:20112,5.1
۳-۱۲ خانواده تجهیز پزشکی
گروهی از تجهیزات پزشکی ساخته شده بوسیله یا برای یک سازمان همسان ، دارای طراحی پایه مشترک و ویژگی های عملکردی یکسان در ایمنی، حیطه کاربرد و کارکرده
۳-۱۳ ارزیابی عملکرد
ارزیابی و تحلیل داده ها برای ایجاد یا تصدیق توانایی یک تجهیز پزشکی تشخیصی در دستیابی به حیطه کاربرد آن .
۳-۱۴ مراقبت پس-بازار
فرایندی نظام مند در گردآوری و تحلیل تجربه به دست آمده از تجهیزات پزشکی که در بازار قرار گرفته اند.
۳-۱۵ محصول
نتیجه یک فرایند
یادآوری: چهار طبقه بندی عمومی از محصولات، شامل موارد زیر وجود دارد:
خدمات (مانند حمل و نقل) و نرم افزار (مانند برنامه کامپیوتری، لغت نامه) • سخت افزار (مانند قطعه مکانیکی موتور) • مواد فرآوری شده مانند روان کننده)
بسیاری از محصولات دربرگیرنده اجزای متعلق به طبقات محصولات عام مختلف ، هستند. این که محصول, خدمت، نرم افزار، سخت افزار یاماده فرآوری شده نامیده شود، بستگی به جزء غالب دارد. به عنوان مثال، محصول ارائه شده «خودرو» از سخت افزار (مانند چرخ ها)، مواد فرآوری شده مانند سوخت، مایع خنک کننده)، نرم افزار (مانند نرم افزار کنترل موتور، کتابچه ی راهنای راننده و خدمت (مانند توضیحات عملکردی داده شده از جانب فروشنده ) تشکیل شده است.
یادآوری: خدمت، نتیجه حداقل یک فعالیت است که ضرورتا در فصل مشترک میان تامین کننده و مشتری انجام شده و عموما ناملموس است. ارائه یک خدمت به عنوان مثال موارد زیر را در بر میگیرد:
- یک فعالیت انجام شده روی محصول ملموس تامین شده از جانب مشتری (مانند اتومبیل که بایستی تعمیر شود)
- یک فعالیت انجام شده روی محصول ناملموس تامین شده از جانب مشتری (مانند ترازنامه درآمدی مورد نیاز برای تهیه اظهارنامه مالیاتی)
- تحویل یک محصول ناملموس (مانند تحویل اطلاعات در بستر انتقال دانش)
- فراهم سازی محیطی برای مشتری (مانند هتل ها و رستوران ها)
نرم افزار از اطلاعات تشکیل شده و عموما ناملموس است و می تواند به اشکال رویکردها، تراکنش هایا روش های اجرائی باشد. |
سخت افزار عموما ملموس بوده و مقدار آن، ویژگی قابل شمارش می باشد. مواد فرآوری شده عموما ملموس بوده و مقدار آنها، ویژگی پیوسته می باشد. سخت افزار و مواد فرآوری شده اغلب بصورت کالا ، عنوان می شوند.
یادآوری: تعریف محصول با تعریف ارائه شده در ISO9000:2015 تفاوت دارد.
۳-۱۶ محصول خریداری شده
محصول ارائه شده از جانب طرفی بیرون از سیستم مدیریت کیفیت سازمان.
یادآوری : ارائه محصول، ضرورتا به ترتیبات تجاری یا مالی منتج نمی گردد .
۳-۱۷ ریسک
ترکیبی از احتمال وقوع یک آسیب و شدت آن آسیب.
یادآوری: تعریف ریسک با تعریف ارائه شده در ISO9000:2015
تفاوت دارد. منبع : ISO14971:2007,2016
۳-۱۸ مدیریت ریسک
به کارگیری نظامند خط مشی های مدیریتی،
رویه ها و شیوه ها در فعالیت های تحلیل، ارزیابی، کنترل و پایش خطر. منبع : ISO14971:2007,2.22
۳-۱۹ سیستم حائل سترونی
حداقل بسته بندی که از ورود ریز جانداران جلوگیری کرده و اجازه حضور محصولی ضد عفونی شده را در هنگام مصرف می دهد. منبع : ISO11607:2006,3,22
۳-۲۰ تجهیز پزشکی سترون
تجهیز پزشکی که مد نظر است الزامات سترونی را برآورده کند.
یادآوری: الزامات جهت سترونی تجهیزات پزشکی ، میتواند موضوعی برای الزامات قانونی اجرائی یا استاندارد ها باشد.
۴- سیستم مدیریت کیفیت
۴-۱ الزامات عمومی ۴-۱-۱
سازمان باید سیستم مدیریت کیفیتی را مدون کرده و اثربخشی آن را در انطباق با الزامات این استاندارد بین المللی و الزامات قانونی اجرائی حفظ نماید.
سازمان باید هر گونه الزام، روش اجرائی، فعالیت یا تمهیدی را که برای مدون سازی در این استاندارد بین المللی یا الزامات قانونی اجرائی مورد نیاز است ، استقرار داده، پیاده سازی کرده و حفظ کند.
سازمان باید نقش به عهده گرفته شده از جانب خود را تحت الزامات قانونی اجرائی ، مدون نماید.
یادآوری: نقش به عهده گرفته شده می تواند شامل سازنده، نماینده مجاز، وارد کننده یا توزیع کننده باشد.
۴-۱-۲ سازمان باید:
أ) فرایندهای موردنیاز برای سیستم مدیریت کیفیت و کاربرد این فرایندها را در سراسر سازمان با در نظر گرفتن نقش به عهده گرفته شده از جانب خود، شناسایی نماید.
ب) یک رویکرد مبتنی بر ریسک را جهت کنترل فرایندهای مناسب موردنیاز در سیستم مدیریت کیفیت، به کار بندد.
پ) توالی و تعامل این فرایندها را تعیین نماید.
۴-۱-۳ سازمان باید برای هر فرایند سیستم مدیریت کیفیت:
أ) شاخص ها و روش های مورد نیاز را جهت حصول اطمینان از اثربخش بودن اجرا و کنترل این فرایندها، مشخص کند
ب) از در دسترس بودن منابع و اطلاعات ضروری برای پشتیبانی از اجرا و پایش این فرایندها, اطمینان حاصل نماید
پ) اقدامات ضروری جهت دستیابی به نتایج طرح ریزی شده و حفظ اثر بخشی این فرایندها را به اجرا گذارد
ت) این فرایندها را پایش, در موارد مقتضی اندازه گیری و تحلیل نماید
ث) سوابق موردنیاز برای اثبات انطباق با این استاندارد بین المللی و الزامات قانونی اجرائی را ثبت کرده و نگهداری نماید ۴-۱-۴
سازمان باید فرایندهای سیستم مدیریت کیفیت را برطبق الزامات این استاندارد بین المللی و الزامات قانونی اجرائی ، مدیریت نماید. تغییرات انجام شده روی این فرایندها باید:
أ) از نظر اثرگذاری بر سیستم مدیریت کیفیت، ارزیابی شوند
ب) از نظر تاثیرگذاری بر روی تجهیز پزشکی تولید شده تحت این سیستم مدیریت کیفیت، ارزیابی شوند
پ) بر طبق الزامات این استاندارد بین المللی و الزامات قانونی اجرائی ، کنترل شوند
۴-۱-۵ زمانی که سازمان تصمیم به برونسپاری
هر گونه فرایند تاثیرگذار بر انطباق محصول با الزامات میگیرد. این برونسپاری باید پایش شده و از کنترل بر روی این گونه فرایندها، اطمینان حاصل شود. سازمان باید مسئولیت انطباق با این استاندارد بین المللی ، الزامات مشتری و الزامات قانونی اجرائی را در مورد فرایندهای برونسپاری شده، به عهده بگیرد. کنترل ها باید متناسب با ریسک های دخیل شده و توانایی طرف بیرونی در برآورده سازی الزامات بر طبق بند ۷-۴) باشد. کنترل ها باید شامل توافقات کیفیتی مکتوب باشد.
۴-۱-۶ سازمان باید روش اجرائی برای صحه گذاری برنامه های نرم افزاری کامپیوتری مورد استفاده در سیستم مدیریت کیفیت مدون کند. این گونه برنامه های نرم افزاری باید پیش از نخستین استفاده و در موارد مقتضی، پس از تغییرات در اینگونه نرم افزارهایا برنامه آنها صحه گذاری شود.
رویکرد و فعالیت های خاص مربوط به صحه گذاری و صحه گذاری مجدد نرم افزار باید با ریسک های مرتبط با کاربری آن متناسب باشد.
سوابق چنین فعالیت هایی باید نگهداری ش ود. ۵-۲-۴ را ببینید).
۴-۲ الزامات مستندسازی
۴-۲-۱ کلیات
مستند سازی سیستم مدیریت کیفیت (424 را ببینید) باید شامل موارد زیر باشد:
آ) بیانیه های مدون از خط مشی کیفیت و اهداف کیفیت
ب) یک نظامنامه کیفیت
پ) روش های اجرائی مدون و سوابق مورد نیاز در این استاندارد بین المللی
ات) مدارک ، شامل سوابقی که از سوی سازمان برای اطمینان از طرح ریزی, اجرا و کنترل موثر فرایندها، ضروری می باشد
ث) هر گونه مستندات مشخص شده در الزامات
قانونی اجرائی ۴-۲-۲
نظامنامه کیفیت سازمان باید یک نظامنامه کیفیت را که شامل موارد زیر است، مدون نماید:
آ) دامنه شمول سیستم مدیریت کیفیت شامل جزییات و توجیه هرگونه حذف یاعدم کاربرد
ب) روش های اجرائی مدون برای سیستم مدیریت کیفیت یا ارجاع به آنها
پ) توصیفی از تعامل فرایندهای سیستم مدیریت کیفیت
نظامنامه ی کیفیت باید ساختار مستندسازی بکار گرفته شده در سیستم مدیریت کیفیت را به طور اجمالی بیان نماید.
۴-۲-۳ پرونده فنی
سازمان باید یک یا چند پرونده را که شامل مدارک تولید شده و یا ارجاع داده شده برای اثبات انطباق با الزامات این استاندارد بین المللی و تطابق با الزامات قانونی اجرائی بوده ، برای هر نوع تجهیز پزشکی یا خانواده تجهیز پزشکی ایجاد و نگهداری نماید.
محتوای پرونده (ها) باید شامل موارد زیر باشد، ولی محدود به آنها نگردد: |
أ) توصیف کلی تجهیز پزشکی ، حیطه کاربرد و برچسب گذاری شامل هرگونه دستورالعمل برای استفاده
ب) مشخصات محصول پ) مشخصات یا روش های اجرائی برای
ساخت، بسته بندی، انبارش، جابجایی و توزیع
ت) روش های اجرائی برای پایش و اندازه گیری
ث) در موارد مقتضی ، الزامات برای نصب
ج) در موارد مقتضی، روش های اجرائی برای خدمات دهی
۴-۲-۴ کنترل مدارک
مدارک الزام شده توسط سیستم مدیریت کیفیت باید کنترل شوند. سوابق , نوع ویژه ای از مستندات هستند و باید مطابق با الزامات ارائه شده در بند 5-2-4 کنترل شوند.
روش اجرائی مدونی باید کنترل های مورد نیاز برای موارد زیر را تعیین نماید:
أ) بازنگری و تصویب مدارک از نظر کفایت پیش از صدور
ب) بازنگری و به روزرسانی، در صورت نیاز، و تصویب مجدد مدارک
پ) اطمینان از این که وضعیت بازنگری کنونی و تغییرات مدارک شناسایی شده است
ت) اطمینان از این که ویرایش های مرتبط از مدارک کاربردی , در محل های استفاده در دسترس می باشد
ث) اطمینان از این که مدارک خوانا و قابل تشخیص باقی می مانند
ج) اطمینان از این که مدارک با منشاء برون سازمانی، که توسط سازمان برای اجرا و طرح ریزی سیستم مدیریت کیفیت ضروری تشخیص داده شده است، شناسایی شده و توزیع آنها کنترل می شود
چ) جلوگیری از مفقود شدن یا از بین رفتن مدارک
ح) جلوگیری از استفاده سهوی مدارک منسوخ شده و به کارگیری روشی جهت شناسایی مناسب آنها
سازمان باید اطمینان یابد که تغییرات در مدارک، توسط همان بخش یا بخش هایی که تصویب اولیه را به انجام رسانده اند و یا بخش های تعیین شده ، مورد بازنگری و تصویب قرار می گیرند. بخش های تعیین شده می بایست به سوابق اطلاعات مربوطه که بر مبنای آنها بازنگری و تصویب مدارک انجام گرفته، دسترسی داشته باشد.
سازمان باید دوره زمانی را مشخص کند که در آن حداقل یک نسخه از مدارک منسوخ ، نگهداری شود. این دوره زمانی باید اطمینان دهد مدارکی که ابزار پزشکی برطبق آنها تولید و بازرسی شده، حداقل تا پایان عمر مفید ابزار که سازمان تعیین می نماید، در دسترس می باشد (این عمر مفید نباید از زمان نگهداری سوابق مربوطه (بند ۴-۲-۴) یا زمان تعیین شده توسط الزامات قانونی اجرائی کمتر باشد).
۴-۲-۵ کنترل سوابق
سوابق باید جهت فراهم آوردن شواهد انطباق با الزامات و اثربخشی اجرای سیستم مدیریت کیفیت استقرار یافته و نگهداری شوند.
سازمان باید روش اجرائی مدونی به منظور تعریف کنترل های موردنیاز برای شناسایی بایگانی, امنیت و یکپارچگی بازیابی, مدت نگهداری و تعیین تکلیف سوابق ایجاد نماید.
سازمان باید روش هایی را مطابق با الزامات قانونی اجرائی برای حفاظت از اطلاعات محرمانه سلامت موجود در سوابق تعریف و پیاده سازی نماید.
سوابق باید خوانا باقی مانده و به سادگی قابل شناسایی و بازیابی باشند. تغییرات در سوابق باید قابل شناسایی باقی بماند.
سازمان باید سوابق را حداقل برای دوره زمانی برابر با دوره عمر تجهیز پزشکی که توسط سازمان تعریف یا توسط الزامات قانونی اجرائی مشخص شده ، نگهداری نماید. این دوره زمانی نباید کمتر از ۲ سال از تاریخ ترخیص محصول باشد.
۵- مسئولیت مدیریت
۵-۱ تعهد مدیریت
مدیریت ارشد باید شواهدی دال بر تعهد خود در ایجاد و تکوین و اجرای سیستم مدیریت کیفیت و نگهداری مداوم اثر بخشی آن به طرق زیر فراهم آورد:
الف) انتقال و تفهیم اهمیت برآورده کردن خواسته های مشتری وهمچنین الزامات قانونی اجرائی در سازمان
ب) تعیین و برقرار کردن خط مشی کیفیت
ج ) اطمینان از اینکه اهداف کیفیت تعیین شده اند
د) انجام بازنگری های مدیریت ه) اطمینان از در دسترس بودن منابع
۵-۲ مشتری مداری
مدیریت ارشد باید اطمینان یابد که الزامات مشتری و الزامات قانونی اجرائی مشخص شده و برآورده شده اند.
۵-۳ خط مشی کیفیت
مدیریت ارشد باید اطمینان یابد که خط مشی کیفیت:
أ) مناسب و مرتبط با مقاصد سازمان می باشد
ب) شامل تعهد به تطابق با الزامات و نگهداشت اثربخشی سیستم مدیریت کیفیت می باشد
پ) چارچوبی را برای استقرار و بازنگری اهداف کیفیت ارائه می نماید
ت) در درون سازمان اطلاع رسانی و درک شده است
ث) از نظر تداوم مناسب بودن مورد بازنگری قرار می گیرد
۵-۴ طرح ریزی ۵-۴-۱
اهداف کیفیت مدیریت ارشد باید اطمینان یابد که اهداف کیفیت و از جمله آنهایی که جهت برآورده کردن
ISO الزامات قانونی اجرائی و الزامات محصول مورد نیاز هستند، در بخش ها و سطوح مرتبط در درون سازمان تعیین شده اند. اهداف کیفیت باید قابل اندازه گیری بوده و باخط مشی کیفیت سازگار باشند.
۵-۴-۲ طرح ریزی سیستم مدیریت کیفیت
مدیریت ارشد باید اطمینان یابد که :
الف) طرح ریزی سیستم مدیریت کیفیت به منظور برآورده کردن الزامات مذکور در بند۱-۴ و همچنین اهداف کیفیت انجام گرفته است
ب) هنگامی که تغییرات در سیستم مدیریت کیفیت طرح ریزی و اجرا می گردد، انسجام سیستم مدیریت کیفیت برقرار نگهداشته می شود
۵-۵ مسئولیت اختیار و انتقال اطلاعات ۵-۵-۱
مسئولیت و اختیار مدیریت ارشد باید اطمینان یابد که مسولیت ها واختيارات تعيين، مدون ودردرون سازمان اطلاع رسانی می شوند.
مدیریت ارشد، باید ارتباط درونی بین کارکنانیکه کارهای موثر بر کیفیت را اداره، اجرا وتصدیق می کنند تعیین نماید. وهمچنین باید از آزادی عمل و اختیار سازمانی لازم برای انجام چنین فعالیت هایی ، اطمینان حاصل نماید. ۵-۵-۲
نماینده مدیریت مدیریت ارشد باید یکی از مدیران خود را به عنوان نماینده مدیریت منصوب کند که جدا
از سایر مسوولیت هایش ، دارای مسوولیت ها و اختیاراتی شامل موارد زیر باشد:
الف) حصول اطمینان از اینکه فرآیندهای مورد نیاز برای سیستم مدیریت کیفیت مدون شده است
ب ) گزارش دهی به مدیریت ارشد در مورد عملکرد سیستم مدیریت کیفیت وهرنوع نیاز برای بهبود
ج ) حصول اطمینان از افزایش آگاهی درخصوص الزامات قانونی اجرائی و الزامات سیستم مدیریت کیفیت در سرتاسر سازمان
۵-۵-۳ ارتباطات داخلی
مدیریت ارشد باید اطمینان یابد که فرآیندهای مناسب انتقال اطلاعات در درون سازمان ایجاد شده و در راستای اثربخشی سیستم مدیریت کیفیت می باشند.
۵-۶ بازنگری مدیریت ۵-۶-۱
کلیات سازمان باید روش اجرائی برای بازنگری مدیریت مدون نماید. مدیریت ارشد باید سیستم مدیریت کیفیت سازمان را در فواصل طرحریزی شده مدون ، بازنگری کند تا از تداوم مناسب بودن, کفایت و اثربخشی آن اطمینان یابد. این بازنگری باید شامل بازنگری فرصت های بهبود و نیاز به تغییرات در سیستم مدیریت کیفیت از قبیل خط مشی کیفیت و اهداف کیفیت باشد.
سوابق بازنگری مدیریت باید نگهداری شوند
در وندادهای بازنگری در وندادهای بازنگری مدیریت باید شامل اطلاعات زیر، و نه محدود به آنها، باشد:
أ) بازخوردها ب) رسیدگی به شکایات پ) گزارش دهی به نهادهای مقرراتی ت) ممیزی ث) پایش و اندازه گیری فرایندها ج) پایش و اندازه گیری محصول چ) اقدامات اصلاحی ح) اقدامات پیشگیرانه خ) اقدامات پیگیرانه بازنگری مدیریت قبلی ر) تغییراتی تاثیر گذار بر سیستم مدیریت کیفیت ز) توصیه هایی جهت بهبود
د) الزامات مقرراتی قابل کاربرد جدید یا بازنگری شده
۵-۶-۳ بروندادهای بازنگری
بروندادهای بازنگری مدیریت باید ثبت شده و شامل بازنگری در وندادهاو هر گونه تصمیم و اقدامی در ارتباط با موارد زیر باشد:
أ) بهبودهای موردنیاز برای حفظ تناسب، کفایت و اثربخشی سیستم مدیریت کیفیت و فرایندهای آن
ب) بهبود محصول در ارتباط با الزامات مشتری
پ) تغییرات در واکنش به الزامات مقرراتی قابل کاربرد جدید یا بازنگری شده
ISO د) نیازهای مربوط به منابع ۶ مدیریت منابع ۶-۱
تامین منابع سازمان باید منابع موردنیاز برای موارد زیر را، تعیین و فراهم نماید:
أ) پیاده سازی سیستم مدیریت کیفیت و نگهداشت اثربخشی آن
ب) برآورده نمودن الزامات قانونی اجرائی و الزامات مشتری
۶-۲ منابع انسانی
کارکنانی که کارهای تاثیرگذار بر کیفیت محصول انجام می دهند. باید شایستگی های لازم را بر پایه تحصیلات, آموزش مهارت ها و تجارب مناسب داشته باشند.
سازمان باید فرایندهایی را برای استقرار شایستگی، ارائه آموزش لازم و اطمینان از آگاهی کارکنان، مدون کند.
سازمان باید:
أ) شایستگی لازم برای کارکنانی که کارهای تاثیرگذار بر کیفیت محصول انجام می دهند، تعیین کند
ب) آموزش ها و یا اقداماتی را جهت دستیابی به شایستگی های لازم و یا حفظ آنها ، ارائه دهد.
پ) اثربخشی اقدامات صورت پذیرفته را مورد ارزیابی قرار دهد
ت) اطمینان یابد که کارکنانش از ارتباط و اهمیت فعالیت هایشان و چگونگی سهیم بودن
شان در دست یابی به اهداف کیفیت, آگاهی دارند
ث) سوابق مناسب مربوط به تحصیلات آموزش ها, مهارت ها و تجارب را نگهداری کند.
یادآوری: روش مورد استفاده برای ارزیابی اثربخشی ، با ریسک های مرتبط با کاری که برای آن آموزش ارائه شده است ، متناسب باشد.
۶-۳ زیرساخت
سازمان باید الزامات را برای زیرساخت های موردنیاز برای دست یابی به انطباق با الزامات محصول، جلوگیری از درهم شدن محصولات و اطمینان از جابجایی منظم محصول، مدون کند.
زیرساخت ، در موارد مقتضی، شامل موارد زیر است: أ) ساختمان ها, فضای کاری و امکانات مرتبط ب) تجهیزات فرایندی (هم سخت افزار هم نرم افزار)
پ) خدمات پشتیبانی ( نظیر حمل و نقل ارتباطات و سیستم های اطلاعاتی)
سازمان باید الزاماتی را برای فعالیت های نگهداری ، شامل تناوب اجرای آنها در مواقعی که این گونه فعالیت هایانبود آنها بتواند بر
کیفیت محصول تاثیرگذار باشد، مدون نماید. در موارد مقتضی، الزامات باید برای تجهیزات مورد استفاده در تولید، کنترل محیط کاری و پایش و اندازه گیری ، به کار گرفته شود.
سوابق این گونه فعالیت های حفظ و نگهداری باید نگهداری شود. ۵-۲-۴ را ببینید).
۶-۴ محیط کاری و کنترل آلودگی ۶-۴-۱
محیط کاری سازمان باید الزامات محیط کاری موردنیاز برای دست یابی به انطباق با الزامات محصول را مدون نماید.
چنانچه شرایط محیط کاری بتواند اثر نامطلوب بر کیفیت محصول داشته باشد، سازمان باید الزامات محیط کاری و روش اجرائی پایش و کنترل آن را ، مدون نماید.
سازمان باید:
أ) الزامات سلامت, پاکیزگی و پوشش کارکنان را در صورتی که تماس آنها با محصول یا محیط کار بتواند بر عملکرد یا ایمنی تجهیز پزشکی تاثیر بگذارد. مدون نماید
ب) اطمینان یابد که تمامی کارکنانی که ملزم هستند موقتا در محیط کار تحت شرایط محیطی خاصی کار کنند. شایسته بوده و یا از سوی فردی شایسته سرپرستی می شوند
* یادآوری: اطلاعات بیشتر در ISO14644 و ISO14968 یافت می شود. ۶-۴-۲
کنترل آلودگی در موارد مقتضی، سازمان باید ترتیباتی را برای کنترل محصول آلوده یا دارای پتاسیل آلودگی به منظور جلوگیری از آلوده شدن محیط کاری، کارکنان یا محصول، طرحریزی و مدون نماید.
برای تجهیزات پزشکی سترون، سازمان باید الزامات کنترل آلودگی با ریزجانداران یا ذرات جامد را مدون کرده و پاکیزگی الزامی را در طی فرایند مونتاژ یا بسته بندی، حفظ کند.
۷- پدید آوری محصول
۷-۱ طرح ریزی پدید آوری محصول
سازمان باید فرایندهای موردنیاز برای پدیدآوری محصول را طرح ریزی نموده و اجرا نماید. طرح ریزی پدیدآوری محصول باید سازگار با الزامات دیگر فرایندهای سیستم مدیریت کیفیت باشد.
سازمان باید یک یا چند فرایند را در پدیدآوری محصول، بمنظور مدیریت ریسک مدون نماید. سوابق فعالیت های مدیریت ریسک باید نگهداری شوند. (۴-۲-۵ را ببینید).
در طرح ریزی پدیدآوری محصول سازمان باید موارد زیر را در موارد مقتضی، تعیین نماید:
أ) اهداف کیفیت و الزامات مربوط به محصول
ب) نیاز به استقرار فرایندها و مستندات ( ۴-۲-۴ را ببینید) و فراهم آوری منابع مختص محصول شامل زیرساخت ها و محیط کاری.
پ) فعالیت های الزامی تصدیق , صحه گذاری پایش اندازه گیری، بازرسی و آزمون, جابجایی، انبارش، توزیع و ردیابی مختص محصول به همراه معیارهای پذیرش محصول
ت) سوابق مورد نیاز جهت فراهم آوردن شواهدی حاکی از اینکه فرآیندهای پدیدآوری و محصول حاصله، الزامات را برآورده می کنند. ( ۴-۲-۵ را ببینید).
برونداد این طرح ریزی باید به صورتی که برای روش های بکار گرفته شده در سازمان مناسب باشد، تدوین گردد. * یادآوری: اطلاعات بیشتر در ISO14971 یافت می شود.
۲-۲ فرایندهای مرتبط با مشتری ۷-۲-۱
تعیین الزامات مرتبط با محصول سازمان باید موارد زیر را مشخص نماید: |
أ) الزامات مشخص شده توسط مشتری شامل الزامات مربوط به فعالیت های تحویل و پس از تحویل
ب) الزامات بیان نشده توسط مشتری که برای استفاده مشخص شده یا حیطه کاربرد (همانگونه که شناخته شده است ) ضروری هستند.
پ) الزامات قانونی اجرائی مرتبط با محصول
ت) هرگونه آموزش موردنیاز کاربر، برای اطمینان از عملکرد مشخص شده و استفاده ایمن از تجهیز پزشکی
ت) هر نوع الزامات دیگر تعیین شده توسط سازمان
۷-۲-۲ بازنگری الزامات مربوط با محصول
سازمان باید الزامات مرتبط با محصول را بازنگری نماید. این بازنگری باید پیش از تعهد سازمان برای تامین یک محصول برای مشتری باشد ( نظیر ارسال مناقصات, پذیرش قراردادها و سفارش ها, پذیرش تغییرات در قراردادها و سفارش ها) و باید اطمینان دهد که:
أ) الزامات محصول تعریف شده و مکتوب شده اند
ب) الزامات قرارداد یا سفارش که با موارد بیان شده درقبل مغایر هستند، حل وفصل شده اند
پ) الزامات مقرراتی قابل کاربرد، برآورده می شوند
ت) هرگونه آموزش موردنیاز کاربر مطابق با ۷-۲-۱ ، فراهم شده یا برنامه ریزی شده است که فراهم گردد
پ) سازمان توانایی برآورده سازی الزامات تعریف شده را دارا می باشد
سوابق نتایج بازنگری و اقدامات حاصله از بازنگری باید نگهداری شود (۵-۲-۴ را ببینید).
هرگاه مشتری خواسته های خود را به صورت مدون بیان نکند، خواسته های مشتری باید پیش از پذیرش بوسیله سازمان مورد تائید قرار گیرد.
هر گاه الزامات و یا خواسته های مربوط به محصول تغییر یابد، سازمان باید اطمینان حاصل کند که مدارک مرتبط اصلاح شده و کارکنان ذیربط از الزامات و یا خواسته های تغییر یافته مطلع گردیده اند.
۷-۲-۳ تبادل اطلاعات
سازمان باید ترتیباتی را برای برقراری ارتباط با مشتریان در ارتباط با موارد زیر طرح ریزی و مدون نماید:
أ) اطلاعات محصول ب) رسیدگی به استعلام ها, قراردادهایا سفارش ها شامل اصلاحیه ها
پ) بازخورد از مشتری شامل شکایات
ت) هشدار های توصیه ای سازمان باید با نهادهای مقرراتی بر طبق الزامات قانونی اجرائی ، تبادل اطلاعات داشته باشد.
۷-۳ طراحی و تکوین ۷-۳-۱
کلیات سازمان باید روش اجرائی برای طراحی و تکوین, مدون نماید.
۷-۳-۲ طرح ریزی طراحی و تکوین
سازمان باید طراحی و تکوین محصول را طرحریزی و تحت کنترل داشته باشد. در موارد مقتضی، مدارک طرحریزی طراحی و تکوین, باید با پیشرفت مراحل طراحی و تکوین, نگهداری شده و به روز رسانی گردد.
در زمان طرحریزی طراحی و تکوین, سازمان باید موارد زیر را مدون نماید:
أ) مراحل طراحی و تکوین
ب) بازنگری های موردنیاز در هر یک از مراحل طراحی و تکوین
پ) فعالیت های تصدیق, صحه گذاری و انتقال طراحی ، مناسب با هر یک از مراحل طراحی و تکوین
پ) مسئولیت ها و اختیارات برای طراحی و تکوین
ت) شیوه های حصول اطمینان از ردیابی بروندادهای طراحی و تکوین به درونداد های طراحی و تکوین
ث) منابع مورد نیاز شامل شایستگی های لازم برای کارکنان
۷-۳-۳ در وندادهای طراحی و تکوین
دروندادهای مرتبط با الزامات محصول, باید تعیین و سوابق آنها نگهداری شود(۴-۲-۴ را ببینید). این در وندادها باید در برگیرنده ی موارد زیر باشد:
أ) الزامات کارکردی, عملکردی, قابلیت استفاده و ایمنی, مطابق با حیطه کاربرد
ب) استانداردها و الزامات قانونی اجرائی پ) برونداد های مناسب با مدیریت ریسک
ت) در موارد مقتضی، اطلاعات به دست آمده از طراحی های مشابه پیشین
ث) دیگر الزامات اساسی برای طراحی و تکوین محصول و فرایندها
این در وندادها باید به لحاظ کفایت و تصویب ، مورد بازنگری قرار گیرند.
الزامات باید کامل و شفاف بوده ، قابلیت تصدیق و صحه گذاری را داشته و در تعارض با یکدیگر نباشند. یادآوری : اطلاعات بیشتر را میتوان درا- IEC62366 یافت نمود
۷-۳-۴ بروندادهای طراحی و تکوین
بروندادهای طراحی و تکوین باید:
أ) الزامات در وندادهای طراحی و تکوین را برآورده نماید
ب) اطلاعات مناسب را برای خرید, تولید و ارائه خدمات فراهم کند
پ) شامل معیارهای پذیرش محصول باشد یا به آنها ارجاع دهد
ت) ویژگی هایی از محصول را که برای استفاده ایمن و مناسب آن ضروری است. مشخص کند.
بروندادهای طراحی و تکوین باید به نحو مناسبی ارایه شوند که بتوان آنها را بر طبق در وندادها طراحی و تکوین تصدیق کرد و باید قبل از ترخیص تایید گردند.
سوابق بروندادهای طراحی و تکوین باید نگهداری شود(۴-۲-۵ را ببینید).
۷-۳-۵ بازنگری طراحی و تکوین در مراحل مناسب, بازنگری های نظام مند طراحی و تکوین باید برطبق ترتیبات طرحریزی شده و مدون، انجام پذیرد تا
أ) توانایی نتایج حاصل از طراحی و تکوین را در برآورده سازی الزامات، ارزیابی کند
ب) هر گونه اقدامات لازم را شناسایی و پیشنهاد کند
شرکت کنندگان در چنین بازنگری هایی باید علاوه بر نمایندگان بخش هایی که با مرحله یا مراحل طراحی و تکوین تحت بازنگری مرتبط هستند، شامل سایر کارکنان متخصص باشند. سوابق نتایج بازنگری ها و هرگونه اقدامات ضروری، باید نگهداری شده و شامل شناسایی طراحی بازنگری شده ، شرکت کنندگان در آن و تاریخ بازنگری باشد(۵-۲-۴ را ببینید).
۷-۳-۶ تصدیق طراحی و تکوین
تصدیق باید بر طبق ترتیبات طرحریزی شده و مدون انجام شود تا اطمینان دهد که بروندادهای طراحی و تکوین الزامات در وندادهای طراحی و تکوین را برآورده می کند.
سازمان باید طرح هائی را جهت تصدیق مدون نماید که شامل روش ها، معیارهای پذیرش و در موارد مقتضی، فنون آماری بر پایه تعیین حجم نمونه باشد.
در صورتیکه حیطه کاربرد محصول الزام نماید که تجهیز پزشکی به دیگر تجهیزیا تجهیزات پزشکی متصل شود یا با آن فصل مشترک داشته باشد، تصديق باید شامل تاییدیه ای باشد که نشان دهد بروندادهای طراحی، در وندادهای طراحی را ، هنگامی که اتصال یا فصل مشترک شکل می گیرد، برآورده می کنند.
سوابق نتایج تصدیق و هرگونه اقدامات ضروری باید نگهداری شود (۴-۲-۴ و ۴-۲-۵ را ببینید).
۷-۳-۷ صحه گذاری طراحی و تکوین
صحه گذاری طراحی و تکوین باید بر طبق ترتیبات طرحریزی شده و مدون انجام شود تا اطمینان حاصل نماید محصول بدست آمده توانائی برآورده سازی الزامات برای کاربرد مشخص شده یا حیطه کاربرد را ، دارد.
سازمان باید طرح هائی را جهت صحه گذاری مدون نماید که شامل روش ها، معیارهای پذیرش و در موارد مقتضی، فنون آماری بر پایه تعیین حجم نمونه باشد.
صحه گذاری طراحی باید بر روی محصول نمونه، انجام شود. محصول نمونه، شامل واحدهای اولیه تولید، بهرهایا معادل آنها، می باشد. منطق بکار گرفته شده برای انتخاب محصول مورد استفاده در صحه گذاری، باید ثبت شود ( ۵-۲-۴ را ببینید).
به عنوان بخشی از صحه گذاری طراحی و تکوین, سازمان باید ارزیابی های بالینی یا ارزیابی عملکرد تجهیز پزشکی را برطبق الزامات قانونی اجرائی انجام دهد. تجهیز پزشکی مورد استفاده برای ارزیابی بالینی یا ارزیابی عملکرد، بعنوان محصول ترخیص شده به منظور استفاده مشتری، تلقی نمی شود.
در صورتیکه حیطه کاربرد محصول الزام کند که تجهیز پزشکی به دیگر تجهیزیا تجهیزات پزشکی متصل شود یا با آن فصل مشترک داشته باشد، صحه گذاری باید شامل تاییدیه ای باشد که نشان دهد الزامات برای کاربرد مشخص شده یا حیطه کاربرد، هنگامی که اتصال یا فصل مشترک شکل می گیرد، برآورده میگردند.
صحه گذاری باید پیش از تحویل یابه کارگیری محصول، تکمیل گردد.
سوابق نتایج صحه گذاری و هر گونه اقدامات
ضروری باید نگهداری شود. (۴-۲-۵ را ببینید).
۷-۳-۸ انتقال طراحی و تکوین
سازمان باید روش اجرائی برای انتقال بروندادهای طراحی و تکوین به تولید، مدون کند. این روش اجرایی باید اطمینان حاصل نماید که برونداد های طراحی و تکوین پیش از تبدیل شدن به مشخصه های نهایی تولید، تصدیق گردیده و برای تولید مناسب هستند و این قابلیت تولید میتواند الزامات مربوط به محصول را برآورده نماید.
سوابق نتایج انتقال و هر گونه اقدامات ضروری باید نگهداری شود. (۴-۲-۵ را ببینید)
۷-۳-۹ کنترل تغییرات طراحی و تکوین
سازمان باید روش اجرائی برای کنترل تغییرات طراحی و تکوین، مدون کند. سازمان باید اهمیت تغییرات در کارکرد، عملکرد، قابلیت استفاده، ایمنی و الزامات قانونی اجرائی را برای تجهیز پزشکی و حیطه کاربرد آن ، تعیین کند.
تغییرات طراحی و تکوین باید شناسایی شوند. پیش از پیاده سازی، تغییرات باید:
آ) بازنگری شده ب) تصدیق شده پ) در موارد مقتضی، صحه گذاری شده ت) تایید شود
بازنگری تغییرات طراحی و تکوین باید در برگیرنده ارزیابی اثر تغییرات بر اجزای متشکله محصول فرآوری شده یاتازه تحویل داده شده ، در وندادها و بروندادهای مدیریت ریسک و فرایندهای پدیدآوری محصول باشد.
سوابق نتایج تغییرات، بازنگری آنها و هرگونه اقدامات ضروری باید نگهداری شود ( ۵-۲-۴ را ببینید).
۷-۳-۱۰ پرونده های طراحی و تکوین
سازمان باید یک پرونده طراحی و تکوین را برای هر نوع از تجهیز پزشکی یا خانواده تجهیز پزشکی، نگهداری کند. این پرونده باید شامل سوابق اثبات انطباق با الزامات طراحی و تکوین و سوابق تغییرات طراحی و تکوین باشد.
۲-۴ خرید ۷-۴-۱
فرایند خرید سازمان باید روش اجرائی را مدون نماید تا اطمینان یابد محصول خریداری شده با اطلاعات مشخص شده خریدر مطابقت دارد.
سازمان باید معیارهایی برای ارزیابی و انتخاب تامین کنندگان ایجاد کند. این معیارها باید بر پایه موارد زیر باشد:
أ) توانایی تامین کنندگان برای ارائه محصولی که الزامات سازمان را برآورده مینماید ،
ب) عملکرد تامین کننده
پ) تاثیر محصول خریداری شده بر کیفیت تجهیز پزشکی
ت) مناسب بودن با ریسک های مرتبط با
تجهیز پزشکی
سازمان باید پایش و ارزیابی مجدد تامین کنندگان را طرحریزی کند. عملکرد تامین کننده باید در برآورده کردن الزامات محصول خریداری شده، پایش شود. نتایج پایش باید دروندادی را برای فرایند ارزیابی مجدد تامین کننده، فراهم کند.
عدم تحقق الزامات خرید، باید تناسب تامین کننده با ریسک مرتبط با محصول خریداری شده و تطابق با الزامات قانونی اجرائی را ، ارجاع دهد.
سوابق نتایج ارزیابی، انتخاب، پایش و ارزیابی مجدد قابلیت یا عملکرد تامین کننده و هرگونه اقدامات ضروری به دست آمده از این فعالیت هار باید نگهداری شود (۴-۲-۵ را ببینید). |
۷-۴-۲ اطلاعات خرید
اطلاعات خرید باید محصولی را که قرار است خریداری شود، توصیف کند یا به آن ارجاع دهد و در موارد مقتضی شامل موارد زیر باشد:
أ) مشخصات محصول
ب) الزامات مربوط به پذیرش محصول, روش اجرائی و فرایندها و تجهیزات
پ) الزامات مربوط به صلاحیت کارکنان تامین کنند
ت) الزامات سیستم مدیریت کیفیت
سازمان باید از کفایت الزامات مشخص شده خرید پیش از اطلاع به تامین کننده, اطمینان یابد
الزامات خرید باید شامل توافق مکتوبی باشد که تامین کننده موظف گردد تغییرات در محصول خریداری شده را به سازمان اطلاع دهد، پیش از آنکه هرگونه تغییر تاثیرگذار بر توانایی محصول خریداری شده در برآورده سازی الزامات خرید را پیاده سازی نماید.
سازمان باید اطلاعات مرتبط با خرید را برای گستره ای از ردیابی مذکور در ۹-۵-۷، به شکل مستندات (۴-۲-۴ را ببینید) وسوابق (۴-۲-۵ را ببینید)، نگهداری نماید.
۷-۴-۳ تصدیق محصول خریداری شده
سازمان باید برای اطمینان از این که محصول خریداری شده الزامات مشخص شده خرید را برآورده می کند. بازرسی یا دیگر فعالیت های ضروری را مستقر و پیاده سازی نماید. گستره فعالیت های تصدیق باید بر پایه نتایج ارزیابی تامین کننده و متناسب با ریسک های مرتبط با محصول خریداری شده باشد.
هنگامی که سازمان از هرگونه تغییری در محصول خریداری شده، آگاه می شود، باید نحوه تاثیر گذاری این تغییرات بر فرایندهای پدیدآوری محصول یا تجهیز پزشکی را تعیین نماید.
هرگاه سازمان یا مشتری آن قصد داشته باشد که تصدیق را در محل های تحت اختیار تامین کننده انجام دهد، سازمان باید فعالیت های تصدیق مورد نظر و شیوه ترخیص محصول را در اطلاعات خریدذکر نماید.
سوابق تصديق باید نگهداری شود (۴-۲-۵ را ببینید).
۷-۵ تولید و ارائه خدمت ۷-۵-۱
کنترل تولید و ارائه خدمت تولید و ارائه خدمت باید جهت اطمینان از انطباق محصول با مشخصات تعیین شده ، طرحریزی ، اجرا ، پایش و کنترل شود . در موارد مقتضی، کنترل های تولید باید شامل موارد زیر ، ونه محدود به آنها، باشد:
أ) مدون سازی روش های اجرائی و شیوه های کنترل تولید
ب) صلاحیت زیرساخت ها
پ) پیاده سازی پایش و اندازه گیری مولفه های فرایند و مشخصه های محصول
ت) دسترس پذیری و استفاده از تجهیزات پایش و اندازه گیری
ث) پیاده سازی عملیات معین شده برچسب گذاری و بسته بندی
ج) پیاده سازی فعالیت های ترخیص, تحویل و پس از تحویل
سازمان باید برای هر تجهیز پزشکی یاهر بهر تجهیزات پزشکی، سوابقی (۴-۲-۵) را پیاده سازی و نگهداری نماید که ارائه دهنده گستره ردیابی مشخص شده در ۹-۵-۷ بوده ، میزان ساخته شده و میزان تایید شده برای توزیع را تعیین نماید. سوابق باید تصدیق و تایید شوند.
۷-۵-۲ پاکیزگی محصول
سازمان باید الزامات پاکیزگی محصول یا کنترل آلودگی آن را, مدون نماید چنانچه :
آ) محصول, پیش از سترون سازی یا مصرف , توسط سازمان تمیز شود
ب) محصول بصورت غیر سترون عرضه گردد و باید پیش از سترون سازی یا مصرف, در معرض فرایند تمیز کاری قرار گیرد
پ) محصول نمی تواند پیش از سترون سازی یا مصرف ، تمیز شود و تمیزی در مصرف آن اهمیت دارد
ت) محصول برای مصارف غير سترون عرضه گردد و تمیزی در مصرف آن اهمیت دارد
ث) عوامل فرآوری در طی ساخت ، از محصول جدا شده باشند
چنانچه محصول مطابق بند آیاب تمیز شده باشد, الزامات بند ۱-۴-۶ پیش از فرایند تمیزکاری کاربرد ندارد.
۷-۵-۳ فعالیت های نصب
سازمان در موارد مقتضی، باید الزامات برای نصب تجهیز پزشکی و معیار پذیرش برای تصدیق نصب را مدون کند.
اگر الزامات توافق شده مشتری اجازه دهد که نصب توسط طرف بیرونی غیر از سازمان یا تامین کننده آن انجام شود. سازمان باید الزامات مدونی را برای نصب تجهیز پزشکی و تصديق آن فراهم نماید.
سوابق نصب تجهیز پزشکی و تصدیق نصب صورت گرفته توسط سازمان یا تامین کننده آن باید نگهداری شود (۵-۲-۴ را ببینید). ۷-۵-۴
فعالیت های خدمات دهی چنانچه خدمات دهی به تجهیز پزشکی الزام مشخص شده باشد. سازمان باید روش اجرائی خدمات دهی, مواد مرجع و اندازه گیری های مرجع را ، در صورت لزوم ، برای انجام فعالیت های خدمات دهی و تصدیق این که الزامات محصول برآورده می شوند، مدون کند.
سازمان باید سوابق فعالیت های خدمات دهی انجام شده توسط سازمان و یا تامین کننده آن را تحلیل نماید تا :
أ) اطلاعاتی که همانند یک شکایت مورد رسیدگی قرار گرفته اند را ، مشخص نماید
ب) در موارد مقتضی ، به عنوان درونداد برای فرایند بهبود، مورد استفاده قرار دهد
سوابق فعالیت های خدمات دهی انجام شده توسط سازمان یا تامین کننده آن ، باید نگهداری شود( ۵-۲-۴ را ببینید)|
۷-۵-۵ الزامات ویژه برای تجهیزات پزشکی استریل شده
سازمان باید سوابق مولفه های فرایند سترون سازی مورد استفاده برای هر بهر ستورن شده را نگهداری کند(۵-۲-۴ را ببینید). سوابق سترون سازی باید برای هر بهر تولیدی تجهیز پزشکی، قابل ردیابی باشد.
۷-۵-۶ صحه گذاری فرایندهای تولید و ارائه خدمت
سازمان باید هر گونه فرایند تولید و ارائه خدمتی که نتایج برونداد آنها با پایش و اندازه گیری پس از آن تصديق نمی شود یا نمی تواند تصدیق شود و نقایص آن به عنوان یک پیامد، تنها پس از استفاده از محصول یا تحویل شدن خدمت آشکار می گردد، صحه گذاری نماید.
صحه گذاری باید توانایی این گونه فرایندها را در دست یابی پایدار به نتایج طرحریزی شده , اثبات نماید.
سازمان باید روش اجرائی برای صحه گذاری فرایندهاشامل موارد زیر مدون کند:
أ) شاخص های تعریف شده برای بازنگری و تایید فرایندها
ب) صلاحیت تجهیزات و کارکنان
پ) استفاده از شیوه ها، روش های اجرائی و معیارهای پذیرش معین
ت) در موارد مقتضی، فنون آماری برپایه تعیین حجم نمونه؛
ت) الزامات مربوط به سوابق (۵-۲-۴ را ببینید)
ث) صحه گذاری مجدد شامل معیارهای صحه گذاری مجدد
ج) تایید تغییرات در فرایندها
سازمان باید روش اجرائی برای صحه گذاری به کارگیری نرم افزارهای کامپیوتری مورد استفاده برای تولید و ارائه خدمات، مدون کند. این گونه برنامه های نرم افزاری باید پیش از اولین استفاده و در موارد مقتضی ، پس از تغییرات در این گونه نرم افزار یا برنامه های آن, صحه گذاری شوند . فعالیت ها و رویکرد مشخص مرتبط با صحه گذاری و صحه گذاری مجدد نرم افزار، باید متناسب با ریسک های مرتبط با استفاده از این نرم افزار شامل تاثیر بر توانایی محصول در انطباق با مشخصات، باشد.
سوابق نتایج و هرگونه اقدام ضروری حاصل از صحه گذاری باید نگهداری شوند(۵-۲-۴ را ببینید).
۷-۵-۷ الزامات ویژه برای صحه گذاری
فرایندهای سترون سازی و سیستم های حائل سترونی سازمان باید روش اجرائی (۴-۲-۴ را ببینید) برای صحه گذاری فرایندهای سترون سازی و سیستم های حائل سترونی, مدون کند.
صحه گذاری این فرایند ها، در موارد مقتضی، باید پیش از پیاده سازی و تغییرات متعاقب محصول یا فرایند, صورت گیرد.
سوابق نتایج و هر گونه اقدام ضروری حاصل از صحه گذاری باید نگهداری شوند (۵-۲-۴ را ببینید). * یادآوری اطلاعات بیشتر را در ISO1l6071 و 2-ISOll607 میتوان یافت.
۷-۵-۸ شناسایی
سازمان باید روش اجرائی برای شناسایی محصول مدون کرده و محصول را از طریق مناسب در سرتاسر پدیدآوری آن، شناسایی نماید.
سازمان باید وضعیت محصول را با توجه به الزامات پایش و اندازه گیری در سرتاسر پدیدآوری محصول شناسایی کند. شناسایی وضعیت محصول باید در سرتاسر تولید انبارش, نصب, یا خدمات دهی محصول حفظ گردد، تا اطمینان حاصل شود تنها محصولی که بازرسی ها و آزمون های الزامی را گذرانده است یا تحت یک اجازه ارفاقی ترخیص شده است، ارسال، استفاده یا نصب می شود.
چنانچه الزامات قانونی اجرائی الزام نماید، سازمان باید سیستمی را برای تخصیص شناسه تجهیز منحصر به فرد برای تجهیز پزشکی، مدون نماید.
سازمان باید روش اجرائی مدون نماید تا اطمینان دهد تجهیزات پزشکی مرجوعی، از دیگر محصولات منطبق شناسایی شده و متمایز میگردد
۷-۵-۹ ردیابی ۷-۵-۹-۱
کلیات سازمان باید روش اجرائی جهت ردیابی مدون نماید. این روش اجرائی باید گستره ردیابی محصول را در مطابقت با الزامات قانونی اجرائی و سوابق نگهداری شده ، تعیین نماید. (۵-۲-۴ را ببینید).
۷-۵-۹-۲ الزامات ویژه برای تجهیزات پزشکی
کاشتنی سوابق الزام شده برای ردیابی باید شامل اجزا, مواد و شرایط محیط کاری مورد استفاده باشد، چنانچه این موارد منجر به این شوند که تجهیز پزشکی، الزامات ایمنی و عملکردی خود را برآورده نکند.
سازمان باید الزام کند که تامین کنندگان توزیع خدمات یا توزیع کنندگان، سوابق توزیع تجهیز پزشکی را جهت بوجود آوردن امکان ردیابی، نگهداری نموده و این سوابق جهت بازرسی در دسترس باشند.
سوابق نام و نشانی دریافت کننده تجهیز پزشکی و باید نگهداری شود(۵-۲-۴ را ببینید).
۷-۵-۱۰ دارایی مشتری
سازمان باید دارایی مشتری را که برای استفاده یا مشارکت در محصول ارائه شده است ، تا هنگامی که تحت کنترل سازمان است یا سازمان از آن استفاده می کند، شناسایی، تصدیق، حفاظت و مراقبت نماید. اگر دارایی مشتری مفقود شده، آسیب دیده، یا به هرنحوی برای استفاده نامناسب باشد، سازمان باید این موارد را به مشتری گزارش داده و سوابق را نگهداری نماید ( ۴-۲-۵ را ببینید). |
۷-۵-۱۱ نگهداری محصول
سازمان باید روش اجرائی برای حفظ انطباق محصول با الزامات، طی فرآوری، انبارش، جابجایی و توزیع، مدون نماید. نگهداری باید بر اجزای تشکیل دهنده یک محصول نیز اعمال شود.
سازمان باید محصول را از دگرگونی ، آلودگی یا
آسیب ، هنگامی که در معرض شرایط و خطرات موردانتظار در طی فرآوری، انبارش، جابجایی و توزیع است، از طریق موارد زیر حفاظت کند:
أ) طراحی و ساخت ظروف بسته بندی و حمل و نقل مناسب
ب) مدون سازی الزامات برای شرایط ویژه موردنیاز، چنانچه بسته بندی به تنهایی نتواند شرایط نگهداری را فراهم نماید
چنانچه شرایط ویژه ای الزام باشد باید کنترل شده و ثبت شود (۵-۲-۴ را ببینید).
۶ کنترل ابزار پایش و اندازه گیری ۷
سازمان باید عملیات پایش و اندازه گیری که قرار است انجام گیرد و همچنین تجهیزات پایش و اندازه گیری مورد نیاز برای فراهم آوری شواهد انطباق محصول با الزامات مشخص شده را تعیین نماید.
سازمان باید روش اجرائی مدون کند تا اطمینان یابد، پایش و اندازه گیری ها می تواند انجام شود و به گونه ای انجام می شود که با الزامات پایش و اندازه گیری سازگار است.
در صورت لزوم به اطمینان از نتایج معتبر، تجهیزات اندازه گیری باید
أ) بوسیله استانداردهای اندازه گیری قابل ردیابی به استانداردهای اندازه گیری بین المللی ياملی، در فواصل زمانی مشخص یا پیش از استفاده، کالیبره یا تصدیق و یا کالیبره و تصدیق
گردد و هر گاه چنین استانداردهایی موجود نباشد، مبنای مورد استفاده برای کالیبراسیون یا تصدیق، باید ثبت گردد.( ۵-۲-۴ را ببینید)
ب) برحسب لزوم تنظیم شده یا تنظیم مجدد گردد. این گونه تنظیم یا تنظیم مجدد ، باید ثبت گردد (۵-۲-۴ را ببینید).
پ) دارای شناسه ای به منظور تعیین وضعیت کالیبراسیون باشد.
ت) از تنظیم هایی که می تواند نتیجه اندازه گیری را نامعتبر سازد مصون نگهداشته شود.
ث) از آسیب دیدگی یا دگرگونی در حین جابجایی، نگهداری یا انبارش مراقبت گردد.
سازمان باید کالیبراسیون یا تصدیق را بر طبق روش اجرائی مدون انجام دهد.
بعلاوه، هرگاه مشخص شود که تجهیزات بکار رفته با الزامات انطباق ندارد سازمان باید اعتبار نتایج اندازه گیری قبلی را ارزیابی و ثبت نماید. سازمان باید اقدام مناسب در مورد تجهیزات و هر نوع محصول تحت تاثیر قرار گرفته را انجام دهد.
سوابق نتایج کالیبراسیون و تصدیق باید نگهداری شود (بند ۵-۲-۴).
سازمان باید روش اجرائی برای صحه گذاری برنامه های نرم افزاری کامپیوتری مورد استفاده برای پایش و اندازه گیری الزامات، مدون کند. این گونه برنامه های نرم افزاری باید پیش از اولین استفاده و در موارد مقتضی ، پس از تغییرات در این گونه نرم افزار یا برنامه های آن , صحه گذاری شوند. فعالیت ها و رویکرد مشخص مرتبط
با صحه گذاری و صحه گذاری مجدد نرم افزار، باید متناسب با ریسک های مرتبط با استفاده از این نرم افزار شامل تاثیر بر توانایی محصول در انطباق با مشخصات باشد.
سوابق نتایج و اقدامات ضروری صحه گذاری باید نگهداری شود (۵-۲-۴ را ببینید). |
یادآوری : اطلاعات بیشتر در ISO10012 یافت می شود.
۸ اندازه گیری, تحلیل و بهبود
۸-۱ کلیات
سازمان باید فرایندهای پایش, اندازه گیری تحلیل و بهبود مورد نیاز برای موارد زیر را ، طرحریزی و به اجرا درآورد :
أ) اثبات انطباق محصول
ب) حصول اطمینان از انطباق سیستم مدیریت کیفیت
پ) حفظ اثربخشی سیستم مدیریت کیفیت
این مورد باید شامل تعیین روش های مقتضی شامل تکنیک های آماری و گستره ی کاربرد آنها با شد ۸-۲
پایش و اندازه گیری ۸-۲-۱
بازخورد سازمان باید اطلاعات مربوط به براورده شدن خواسته های مشتری توسط سازمان را به عنوان یکی از موارد سنجش اثربخشی سیستم مدیریت کیفیت, جمع آوری و پایش نماید. روش دست یابی و استفاده از این اطلاعات باید تدوین شود.
سازمان باید روش اجرائی برای فرایند بازخورد، مدون نماید. فرایند بازخورد باید شامل امکانات گردآوری اطلاعات از فعالیت های تولید و پس از تولید باشد.
اطلاعات گردآوری شده از فرایند بازخورد باید به عنوان درونداد احتمالی مدیریت ریسک برای پایش و اندازه گیری الزامات محصول و نیز پدیدآوری محصول و بهبود مستمر، به خدمت گرفته شود.
چنانچه الزامات مقرراتی، سازمان را ملزم به کسب تجربیاتی از فعالیت های پس از تولید کرده باشد، بازنگری این تجارب، باید بخشی از این فرایند بازخورد باشد.
۸-۲-۲ رسیدگی به شکایت
سازمان باید روش اجرائی برای رسیدگی به موقع به شکایت بر طبق الزامات قانونی اجرائی، مدون نماید.
روش اجرائی باید حداقل شامل الزامات و مسئولیت هایی برای موارد زیر باشد:
أ) دریافت و ثبت اطلاعات
ب) ارزیابی اطلاعات در تعیین مواقعی که بازخورد شامل شکایت گردد
پ) بررسی شکایت
ت) تعیین نیاز به گزارش دهی اطلاعات به نهادهای قانونی ذیصلاح
ث) رسیدگی به محصول مرتبط با شکایت؛ ج) تعیین نیاز به آغاز انجام اصلاحات یا اقدامات اصلاحی
چنانچه هر شکایتی بررسی نشود توجیه این امر باید مدون شود. هر گونه اصلاح یا اقدام اصلاحی حاصل از فرایند رسیدگی به شکایت مشتری، باید مدون گردد.
چنانچه در بررسی یک شکایت ، مشخص شود فعالیت های بیرون از سازمان در شکایت مشارکت داشته اند، اطلاعات مربوطه باید، بین سازمان و طرف بیرونی مرتبط، تبادل گردد .
سوابق رسیدگی به شکایت باید نگهداری شود ( ۴-۲-۵ را ببینید).
۸-۲-۳ گزارش دهی به نهادهای قانونی
چنانچه الزامات قانونی اجرائی، اطلاع رسانی شکایاتی را الزام نماید که شاخص گزارش دهی ویژه رویدادهای نامطلوب یا صدور هشدارهای توصیه ای را دارا میباشند ، سازمان باید روش اجرائی برای ارائه اطلاعات به نهادهای قانونی ذیصلاح، مدون نماید.
سوابق گزارش دهی به نهادهای قانونی باید نگهداری شود (۵-۲-۴ را ببینید).
۸-۲-۴ ممیزی داخلی
سازمان باید ممیزی داخلی را در فواصل زمانی برنامه ریزی شده اجرا نماید تا تعیین کند که آیا سیستم مدیریت کیفیت:
أ) با ترتیبات برنامه ریزی شده و مدون، الزامات این استاندارد بین المللی و الزامات سیستم
مدیریت کیفیت استقرار یافته توسط سازمان و الزامات قانونی اجرائی، انطباق دارد
ب) به طور اثربخش پیاده سازی و نگهداری می شود
سازمان باید یک روش اجرائی برای تشریح مسئولیت ها و الزامات طرحریزی و اجرای ممیزی های داخلی و ثبت و گزارش دهی نتایج ممیزی، مدون نماید.
برنامه ممیزی باید با در نظر گرفتن وضعیت و اهمیت فرآیندها و حوزه هایی که لازم است ممیزی شوند و نیز نتایج ممیزی های قبلی تهیه شود. معیار، دامنه شمول، فواصل زمانی و روش ها ی ممیزی باید تعیین وثبت گردد (۵-۲-۴ راببینید). نحوه انتخاب ممیزان وانجام ممیزی ها باید منجر به حصول اطمینان از عینی بودن و بی طرف بودن فرآیند ممیزی گردد. ممیزان نباید کار خود را ممیزی کنند.
سوابق ممیزی و نتایج آن شامل شناسایی فرایندها و حوزه های ممیزی شده و جمع بندی آنها، باید نگهداری شود (۵-۲-۴)
مدیریت مسئول حوزه تحت ممیزی، باید اطمینان یابد که اصلاحات و اقدامات اصلاحی ضروری برای رفع عدم انطباق های کشف شده و علل آنها، بدون تاخیر صورت پذیرفته است. فعالیتهای پیگیری باید شامل تصدیق اقدامات انجام پذیرفته و گزارش دهی در مورد نتایج تصدیق باشد (بند ۲-۵-۸).
یادآوری : اطلاعات بیشتر در 19011 ISO یافت می شود.
۸-۲-۵ پایش و اندازه گیری فرایندها |
سازمان باید روشهای مناسبی را برای پایش و در موارد مقتضی، اندازه گیری فرآیندهای سیستم مدیریت کیفیت به کار گیرد. این روش ها، باید توانایی فرآیندها را در دستیابی به نتایج طرحریزی شده، اثبات نماید. هرگاه نتایج طرحریزی شده حاصل نگردد، در موارد مقتضی، اصلاحات و اقدامات اصلاحی لازم باید انجام گیرند.
۸-۲-۶ پایش و اندازه گیری محصول
سازمان باید ویژگی های محصول را جهت تصدیق اینکه الزامات و یا خواسته های مربوط به محصول برآورده شده اند مورد پایش و اندازه گیری قرار دهد. این امر باید در مراحل کاربردی از فرآیند پدیدآوری محصول بر طبق ترتیبات مدون و طرح ریزی شده و روشهای اجرایی مدون انجام گیرد.
شواهد انطباق با معیار پذیرش باید نگهداری شود. هویت فرد دارای اختیار برای ترخیص محصول باید ثبت گردد (۵-۲-۴ را ببینید). در موارد مقتضی، سوابق باید تجهیزات آزمون مورد استفاده برای انجام فعالیت های اندازه گیری را شناسایی کند.
ترخیص محصول و ارایه خدمت تا هنگامی که ترتیبات طرح ریزی شده و مدون بطور رضایت بخش تکمیل نشده باشد، نباید صورت گیرد.
برای تجهیزات پزشکی کاشتنی، سازمان باید هویت فردی که هر گونه بازرسی و آزمون را انجام میدهد، ثبت کند.
۸-۳ کنترل محصول نامنطبق ۸-۳-۱
کلیات سازمان باید اطمینان یابد محصولی که با الزامات و یا خواسته های مربوط به آن منطبق نیست بمنظور جلوگیری از استفاده یا تحویل ناخواسته آن شناسایی شده وتحت کنترل می باشد. سازمان باید روش اجرائی برای تعریف کنترل ها و مسئولیت ها و اختیارات مرتبط برای شناسایی، مستندسازی، جداسازی، ارزیابی و تغییر وضعیت محصول نامنطبق مدون کند.
ارزیابی محصول نامنطبق باید شامل تعیین نیاز به یک بررسی و اطلاع رسانی به طرف بیرونی مسئول در قبال عدم انطباق باشد.
سوابق ماهیت عدم انطباق و هرگونه اقدام بعدی انجام شده شامل ارزیابی، هرگونه بررسی و دلائل برای تصمیمات، باید نگهداری شود ( ۴-۲-۵ را ببینید).
۸-۳-۲ اقدام در قبال محصول نامنطبق شناسائی شده پیش از تحویل
سازمان باید با محصول نامنطبق بایک یا چند روش زیر برخورد کند:
أ) انجام اقدامی برای رفع عدم انطباق های تشخیص داده شده
ب) انجام اقدامی برای جلوگیری از بکارگیری یا استفاده اصلی محصول
پ) کسب اختیار برای استفاده, ترخیص یا پذیرش تحت مجوز ارفاقی
سازمان باید اطمینان حاصل کند که محصول نامنطبق تنها زمانی با مجوز ارفاقی پذیرفته می شود که توجیه آن ارائه شده، تاییدیه ها گرفته شده و الزامات قانونی اجرائی برآورده شده باشند . سوابق پذیرش بوسیله اجازه ارفاقی و هویت فرد مجاز برای اعطاء اجازه ارفاقی و باید نگهداری شود (۵-۲-۴ را ببینید).
۸-۳-۳ اقدام در قبال محصول نامنطبق شناسائی شده پس از تحویل
زمانی که محصول نامنطبق پس از تحویل یاشروع استفاده شناسایی گردد , سازمان باید اقداماتی متناسب با آثار یا آثار بالقوه عدم انطباق , انجام دهد. سوابق اقدامات انجام شده باید نگهداری شود (۵-۲-۴ را ببینید).
سازمان باید روش اجرائی برای صدور هشدارهای توصیه ای برطبق الزامات قانونی اجرائی، مدون نماید. این روش اجرائی باید قابلیت اجرا در هر زمانی را داشته باشد. سوابق اقدامات مربوط به صدور هشدارهای توصیه ای باید نگهداری شود ( ۴-۲-۵ را ببینید). |
۸-۳-۴ باز کاری
سازمان باید باز کاری را بر طبق روش اجرائی
مدون که آثار نامطلوب بالقوه باز کاری را بر روی محصول در نظر میگیرد، اجرا نماید. این روش اجرائی باید تحت همان بازنگری و تاییدیه روش اصلی باشد.
پس از اتمام بازکاری، محصول باید تصدیق شود تا اطمینان دهد شاخص های پذیرش کاربردی و الزامات قانونی برآورده می شود .
سوابق باز کاری باید نگهداری شود. ۵-۲-۴ را ببینید).
۸-۴ تحلیل داده ها
سازمان باید روش اجرائی برای تعیین , گردآوری و تحلیل داده های مناسب مدون نماید.، تاسازگاری ، کفایت و اثربخشی سیستم مدیریت کیفیت، را نشان دهد.
روش اجرائی باید شامل تعیین روش های مناسب، مانند فنون آماری و گستره به کارگیری آنها باشد.
تحلیل داده باید شامل داده های حاصله به عنوان نتیجه پایش و اندازه گیری و دیگر منابع مرتبط بوده و حداقل شامل ورودی هایی از موارد زیر باشد:
آ) بازخورد ب) انطباق با الزامات محصول
پ) ویژگی ها و روند فرایندها و محصولات شامل فرصت هایی برای بهبود
ت) تامین کنندگان ث) ممیزی ها | ج) گزارش های خدمات دهی ، در موارد مقتضی
چنانچه تحلیل داده ها نشان دهد که سیستم مدیریت کیفیت ، مناسب ، کافی و موثر نمی باشد سازمان باید از این تحلیل ها بعنوان ورودی بهبود مورد نیاز در ۸-۵ استفاده نماید.
سوابق نتایج تحلیل ها باید نگهداری شوند ( ۴-۲-۵ را ببینید).
۸-۵ بهبود ۸-۵-۱
کلیات سازمان باید هر گونه تغییر ضروری را شناسایی و پیاده سازی کند تا از تداوم سازگاری، کفایت و اثربخشی سیستم مدیریت کیفیت و ایمنی و عملکرد تجهیز پزشکی از طریق استفاده از خط مشی کیفیت، اهداف کیفیت، نتایج ممیزی ها، مراقبت پس بازار، تحلیل داده ها، اقدامات اصلاحی، اقدامات پیشگیرانه و بازنگری مدیریت، اطمینان یافته و آن را نگهداری نماید.
۸-۵-۲ اقدام اصلاحی
سازمان باید اقداماتی را برای رفع علت عدم انطباق ها, به منظور جلوگیری از وقوع مجدد آنها به انجام رساند. هرگونه اقدام اصلاحی ضروری باید بدون هرگونه تاخیری صورت گیرد. اقدام اصلاحی باید متناسب با اثرات عدم انطباق های بوقوع پیوسته باشد.
سازمان باید یک روش اجرائی مدون نماید تا الزاماتی جهت موارد زیر تعیین نماید:
أ) بازنگری عدم انطباق ها (شامل شکایات) ب) تعیین علل عدم انطباق ها
پ) ارزیابی نیاز به اقدام جهت حصول اطمینان از اینکه عدم انطباق ها مجددا رخ ندهند
ت) طرحریزی ، تدوین و پیاده سازی اقدام موردنیاز شامل به روزرسانی مستندات در موارد مقتضی
ث) تصدیق این که اقدام اصلاحی اثر نامطلوبی بر توانایی برآورده سازی الزامات قانونی اجرائی یا ایمنی و عملکرد تجهیز پزشکی ندارد
ج) بازنگری اثربخشی اقدامات اصلاحی انجام شده
سوابق نتایج هر گونه بررسی و اقدامات انجام شده باید نگهداری شود (۵-۲-۴ را ببینید).
۸-۵-۳ اقدام پیشگیرانه
سازمان باید اقداماتی را برای رفع علت عدم انطباق های بالقوه به منظور جلوگیری از وقوع آنها به انجام رساند. اقدام پیشگیرانه باید متناسب با اثرات مشکلات بالقوه باشد.
سازمان باید یک روش اجرائی مدون کند تا الزاماتی جهت موارد زیر تعیین نماید
أ) تعيين عدم انطباق های بالقوه و علل بروز آنها
ب) ارزیابی نیاز به اقدام برای جلوگیری از وقوع عدم انطباق ها
پ) طرحریزی ، تدوین و پیاده سازی اقدام موردنیاز شامل به روز رسانی مستندات در موارد مقتضی
ت) تصدیق این که اقدام پیشگیرانه، اثر نامطلوبی بر توانایی برآورده سازی الزامات قانونی اجرائی یا ایمنی و عملکرد تجهیز پزشکی ندارد؛
ث) بازنگری اثربخشی اقدامات پیشگیرانه انجام شده، در موارد مقتضی
سوابق نتایج هرگونه بررسی و اقدامات انجام شده باید نگهداری شود (۵-۲-۴ را ببینید).


 در حالی که پمپ های تزریق اختراع جدیدی هستند، درمان داخل وریدی در قرون وسطی آغاز شد. اولین دستگاه تزریق با موفقیت توسط دانشمند آکسفورد آقای کریستوفر رن در سال 1656 از مثانه خوک و یک قلم پر ساخته شد. در حالی که موفق بود، رن متوجه شد که دستگاه دوام ندارد و ایمن کردن آن دشوار است. به همین دلیل رن به عنوان پدر درمان داخل وریدی شهرت پیدا کرد.
در حالی که پمپ های تزریق اختراع جدیدی هستند، درمان داخل وریدی در قرون وسطی آغاز شد. اولین دستگاه تزریق با موفقیت توسط دانشمند آکسفورد آقای کریستوفر رن در سال 1656 از مثانه خوک و یک قلم پر ساخته شد. در حالی که موفق بود، رن متوجه شد که دستگاه دوام ندارد و ایمن کردن آن دشوار است. به همین دلیل رن به عنوان پدر درمان داخل وریدی شهرت پیدا کرد.



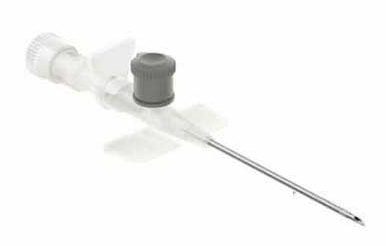
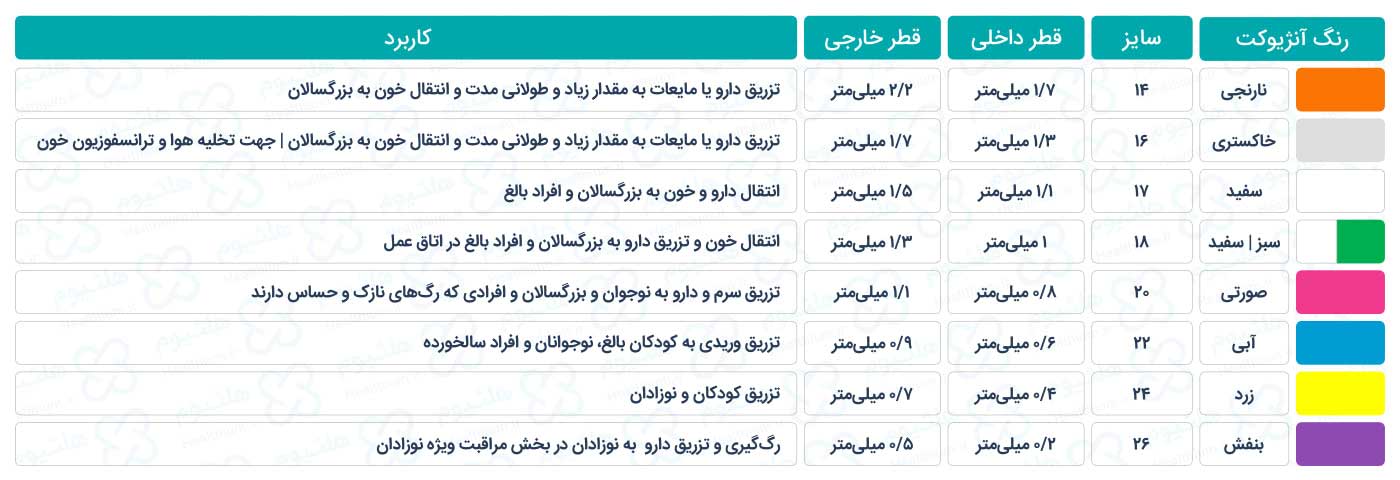
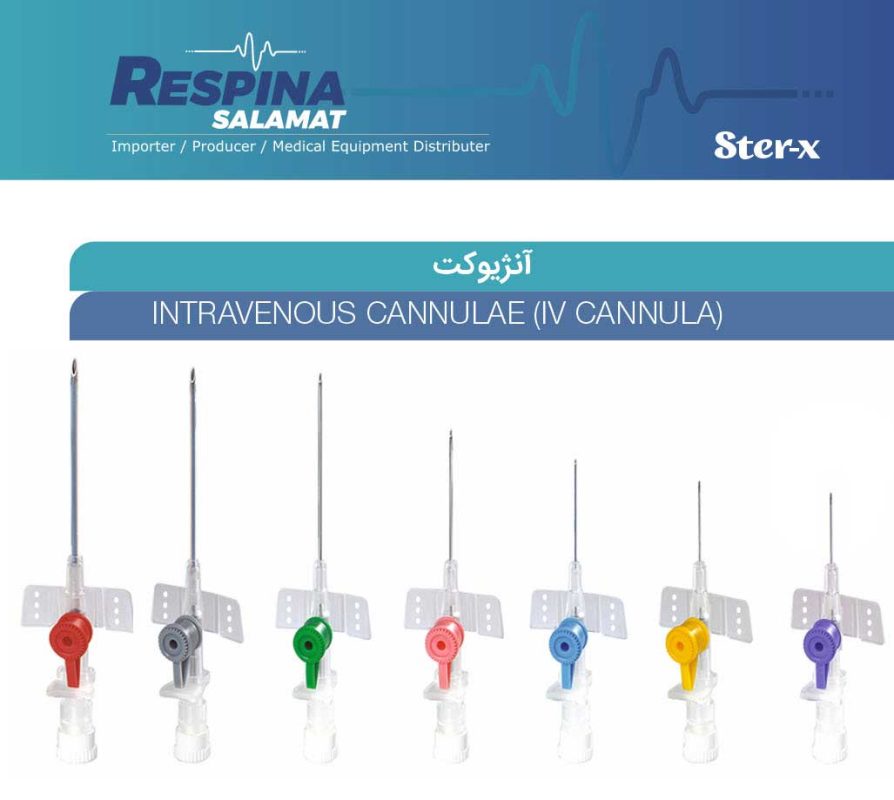
 • آنژیوکت گیج 18
• آنژیوکت گیج 18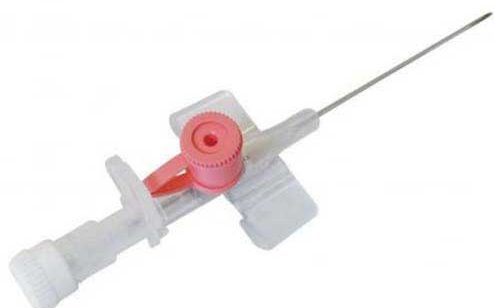 • آنژیوکت گیج 20
• آنژیوکت گیج 20 • آنژیوکت گیج 22
• آنژیوکت گیج 22